
Endocrine disorders in patients with hereditary hemochromatosis


Abstract
Hereditary hemochromatosis (HH) is a rare genetic disorder, developing secondary to the accumulation of iron in tissues, which may lead to multiple organ failure. If untreated, it may result in liver cirrhosis or cardiomyopathy. The damage to the pancreas and the anterior pituitary, on the other hand, leads to a decreased production and secretion of hormones that are essential to life.
Common symptoms of HH, that are distressing for patients, include joint pain, particularly involving hands and wrists, as well as the chronic fatigue syndrome. Iron overload affects the skeletal system, leading to osteoporosis. The pathological accumulation of iron in the anterior pituitary impairs the gonadotropin synthesis, resulting in reduced serum levels of testosterone in men and estrogens in women. This, however, contributes to lower bone mass. In vivo tests have also revealed that abnormal iron accumulation is related to an increased activity and number of osteoclasts, as well as the influence on the differentiation and activity of osteoblast-lineage cells. Based on a systematic review of literature, hereditary hemochromatosis (HH) will be presented as a chronic disease, affecting most of the endocrine glands.
Citation
Banaszkiewicz K, Sikorska K, Sworczak K. Endocrine disorders in patients with hereditary hemochromatosis. Eur J Transl Clin Med. 2018;1(2):72-76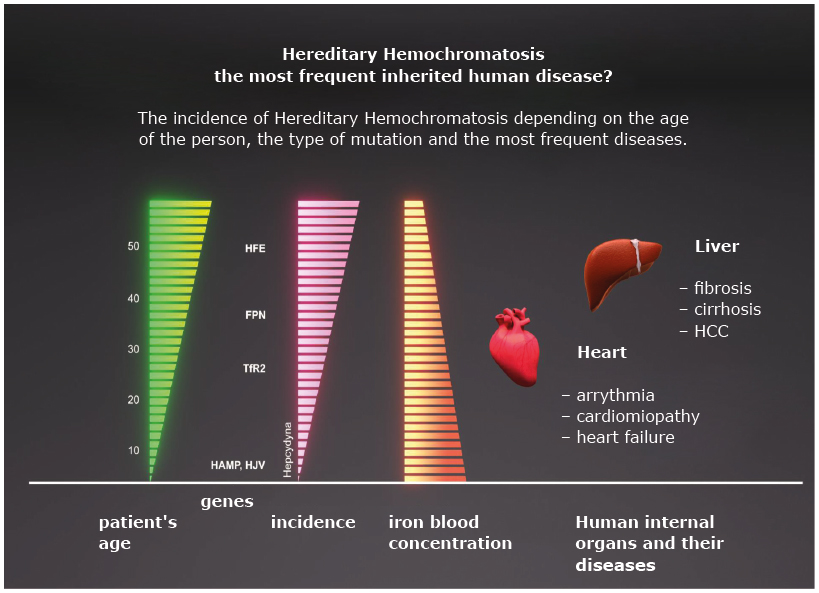

Introduction
HFE (human hemochromatosis protein) – associated hereditary hemochromatosis (HH) is an autosomal recessive a genetic disorder. Among the Caucasian population, mutations of the HFE gene account for 60-96% of cases in which the symptoms of iron overload develop. Classic HH-Hereditary Hemochromatosis develops in homozygotic carriers of the C282Y mutation in the HFE gene – as a result of a point mutation in the nucleotide (845G>A), cysteine is substituted with tyrosine in the HFE protein. The prevalence of this mutation among the Caucasian population has been estimated at 3-10 cases per 1000 people, which means that HH is one of the most prevalent genetic metabolic diseases [1].
Other polymorphisms in the HFE gene (H63D – substitution of histidine with aspartic acid, S65C – substitution of serine with cysteine), which are frequently detected, do not cause such serious alterations within the spatial structure of the HFE protein and such dynamic symptoms of iron overload [2].
Most commonly, the first clinical manifestation of the disease involves symptoms of liver damage, which in most cases of untreated primary HH leads to liver cirrhosis with significant risk of hepatocarcinogenesis [3]. Iron deposition in other organs can also lead to: diabetes mellitus, cardiomyopathy, bone and joint pathology, hypopituitarism and easily noticeable (brownish) skin discoloration. Early diagnosis and treatment aimed at reducing the tissue iron concentration (e.g. bloodletting as a first-line therapy, iron chelation therapy) are of undeniable importance in preventing irreversible organ damage [4-7].
Any pathological changes in endocrine glands may develop secondary to pathological iron accumulation of a different etiology. In classic HH, these changes are usually diagnosed concomitantly with the main liver pathology. In juvenile hemochromatosis, which is a very rare disorder developing secondary to underlying mutations in the hepcidin gene or the hemojuvelin gene with rapidly progressive iron accumulation, hormonal imbalance manifests itself at a younger age in the form of severe endocrinological dysfunctions (Figure 1) [8].
A juvenile hemochromatosis type 2, a disease with severe symptoms of iron accumulation and an early onset, is diagnosed in patients with abnormal synthesis of hepcidin or hemojuvelin. It is a rare disease characterized by a dynamic cycle of iron deposition in tissues, quick depletion of liver’s compensating capacity for storing iron and development of symptoms associated with the failure of the heart muscle and endocrine glands. Mutations in the TFR2 (transferrin receptor 2 gene) encoding gene that cause the dysfunction of this protein rarely lead to hereditary hemochromatosis classified as hemochromatosis type 3. The clinical manifestation of hemochromatosis type 3 is similar to that of hemochromatosis type 1, but the symptoms may develop at a young age [9].
Figure 1. Clinical picture of hemochromatosis depending on mutation of genes encoding HFE, hepcidin (HAMP), hemojuvelin (HJV), transferrin 2 receptor (TfR2), ferroportin (FPN). Organ changes may concern: liver, heart, endocrine organs.
Image modification from: Pietrangelo A.: Hereditary Hemochromatosis: Pathogenesis, Diagnosis and Treatment. Gastroenterology 2010; 139: 393-408
Adrenal gland dysfunction
Hemochromatosis, along with sarcoidosis, amyloidosis and non-Hodgkin lymphomas, is listed as one of the causes of primary adrenal insufficiency, which supposedly develops due to the so-called infiltration of the glandular tissue. According to this hypothesis, excessive accumulation of iron may lead to adrenal damage due to oxidative stress, induced by high oxidation- reduction potential of iron [10].
Numerous data concerning the etiopathogenetic relationship between iron overload and endocrine disease are based on observations of patients with secondary hemochromatosis, e.g. patients diagnosed with thalassemia. Adrenal insufficiency affects 13-46% of such patients. Researchers confirmed the dysfunction of the pituitary gland with a decreased ACTH (adrenocoticotropic hormone) production, but also the dysfunction of the adrenal cortex demonstrated in ACTH stimulation tests [11].
Among few publications devoted to juvenile hemochromatosis, there is a case study from 2000, describing a patient with juvenile hemochromatosis, whose only endocrine disorder was isolated adrenal insufficiency without the presence of anti-adrenal antibodies. The authors suggested that the dysfunction of the adrenal glands in this patient is due to the advanced stage of iron overload in tissues [12]. In 2018 a clinical case was published in which the patient’s adrenal cortex was damaged in the course of hemochromatosis with homozygous H63D mutation, which was confirmed by endocrinological diagnostics and iron deposits in the adrenal MRI examination [13].
Adrenal insufficiency also develops secondary to the dysfunction of the pituitary gland. In the case of hemochromatosis, the increased deposition of iron in the cells of the pituitary gland may lead to a decreased secretion of tropic hormones [14]. The most common endocrine disorder in patients with hereditary hemochromatosis is hypogonadotropic hypogonadism, which causes low libido (26% of patients) and impotence in men (45% of men). Hypogonadism in patients with hemochromatosis is supposedly caused by both the dysfunction of the hypothalamic-pituitary axis and the destruction of Leydig cells [15].
Hypothyroidism
Hypothyroidism is another disorder that can occur among people with congenital hemochromatosis. Published data referring to the incidence of thyroid dysfunctions among patients diagnosed with congenital hemochromatosis are incoherent and inconclusive. In a 1983 study, Edwards and co-authors demonstrated an increased incidence of primary hypothyroidism in patients with congenital hemochromatosis, but more recent reports from 2004 do not confirm this. The explanation for the relatively low incidence of thyroid disease among patients with hereditary hemochromatosis may be the fact that pituitary glandular cells are more sensitive than thyroid cells to the damaging effects of iron deposits [16-17].
Hypogonadism and osteoporosis
Hypogonadism may lead to a decreased bone mass in men, which is a frequent finding in patients with HH. In a study from 2008 conducted on a group of 87 patients with hemochromatosis who were tested for reduced bone mineral density (BMD), it was found that 25% of them suffered from osteoporosis, and 41% from osteopenia. Moreover, it was stated that BMD was dependent on BMI (body mass index), alkaline phosphatase level, hypogonadism/menopause and the intensity of iron deposition in the liver. However, there was no correlation with liver cirrhosis. In a study from 2005 osteopenia was found in 30 out of 38 patients (78.9%), while osteoporosis in 13 out of 38 patients (34%). The decrease in BMD was greater in the femoral neck than in the lumbar region of the spine [18-19].
In vivo tests suggested that there is a relationship between excessive iron accumulation and an increase in the activity and number of osteoclasts. When analyzing the problem, it is also important to remember about the calcium and phosphate homeostasis, which is disturbed in patients with liver impairment. In chronic liver disease, 20-100% of patients develop the so-called hepatic osteodystrophy. It has also been confirmed that iron has a negative influence on bones. In vivo tests revealed that iron overload suppresses osteoblastic differentiation and activity, which is associated with increased apoptosis of osteoblasts [20]. Iron has also a negative impact on bones through BMP2 (Bone Morphogenetic Protein 2) and osteoblastogenesis. Study results from 2009 suggest that iron overload may contribute to the development of osteoporosis as a result of inhibited osteoblastic proliferation and differentiation [21].
In research from 2008, the HFE gene was mutated in rats causing excessive iron accumulation in the kidneys and liver. The research showed that there was a decrease in connections between cells in the trabecular bone of the femur. Recently, the results of the studies analyzing the influence of iron on the microarchitecture of bones have been published. It was found that animals with iron disorders have a reduced BFR (bone formation rate), which suggests that iron has a direct influence on the percentage of active osteoblasts. Based on the bone mass analysis, it was found that animals with iron overload have an impaired microarchitecture of bones due to the compromised formation of the trabecular bone and a decreased number of trabeculae. This mechanism is probably the main cause of the harmful effect of iron on bones [22].
Diabetes mellitus
Decreased insulin secretion is considered the main cause of diabetes in HH patients, with insulin resistance playing the secondary role. Research suggest that bloodletting may delay the development of diabetes type 2 in patients diagnosed with HH. According to the recently published studies, 13-23% of patients with hereditary hemochromatosis suffer from diabetes, a rate much lower than several decades ago [23].
Dubois et al demonstrated that the frequency of the Cys282Tyr and His63Aps mutations of the HFE gene in patients with diabetes type 2 is higher in comparison to the general population. In another article, the same research team investigated whether diabetes type 2 constitutes sufficient evidence to suspect HH and suggested genetic screening only for patients with concomitant liver cirrhosis [24-25].
The strongest correlation between the presence of the HFE gene mutation and the incidence of diabetes type 2 was determined by Moczulski et al. In research from 2001 conducted on a Polish population, the presence of the H63D mutation was found to be a risk factor for diabetic neuropathy, and the prevalence of the C282Y mutation was greater in patients with diabetes type 2 than in the general population [26].
The results of the Hemochromatosis and Iron Overload Screening (HEIRS) study published in 2006 are quite different: in a large group of patients (223 homozygotic carriers of the C282Y mutation and 449 controls; all with no previous history of diabetes), the percentage of patients with undiagnosed diabetes or an abnormal fasting blood glucose level was similar for C282Y homozygotes and the general population [25].
In a study published by Davis et al. in 2008, 1245 patients with diabetes type 2 were evaluated and no correlation was found between increased iron levels, the presence of the HFE gene mutation and diabetes type 2. Based on their results, screening for HH in patients with diabetes is not required [27].
In a meta-analysis of 23 studies conducted between 1997 and 2011, the prevalence of diabetes type 2 in carriers of the C282Y mutation, both homo- and heterozygotic, was comparable to the prevalence of diabetes in the general population. Carriers of the H63D mutation in the HFE gene had a slightly higher risk for developing diabetes type 2 [28].
Conclusions
Taking into consideration the latest research, it is not diabetes that seems to be the most common endocrinopathy in hemochromatosis, but hypogonadism – it does not only lower the quality of life, but may also lead to insidious and gradually progressive osteoporosis, fractures and progressing disability. Patients with HH should be monitored by both a hepatologist and endocrinologist, and a special emphasis should be put on the necessity of performing screening tests for decreased bone mineral density. The treatment should not be reduced only to decreasing the amount of iron in the organism, but it should also include vitamin D3 supplementation, gonadal hormone replacement therapy and the use of bisphosphonates.
References
| 1. |
Sikorska K, Bielawski KP, Romanowski T, Stalke P. Hereditary hemochromatosis: the most frequent inherited human disease. Postepy Hig Med Dosw (Online). 2006;60:667-76.
|
| 2. |
Aranda N, Viteri FE, Montserrat C, Arija V. Effects of C282Y, H63D, and S65C HFE gene mutations, diet, and life-style factors on iron status in a general Mediterranean population from Tarragona, Spain. Ann Hematol. 2010;89(8):767-73.
|
| 3. |
Niederau C, Fischer R, Purschel A, Stremmel W, Haussinger D, Strohmeyer G. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology. 1996;110(4):1107-19.
|
| 4. |
Niederau C, Fischer R, Sonnenberg A, Stremmel W, Trampisch HJ, Strohmeyer G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313(20):1256-62.
|
| 5. |
Beddy P, McCann J, Ahern M, Norris S, Keogan M. MRI assessment of changes in liver iron deposition post-venesection. Eur J Radiol. 2011;80(2):204-7.
|
| 6. |
Niederau C, Stremmel W, Strohmeyer GWW. 7 Clinical spectrum and management of haemochromatosis. Baillieres Clin Haematol. 1994;7(4):881-901.
|
| 7. |
Rozwadowska K, Daniłowicz‐Szymanowicz L, Fijałkowski M, Sikorska K, Gałąska R, Kozłowski D, et al. Can two‐dimensional speckle tracking echocardiography be useful for left ventricular assessment in the early stages of hereditary haemochromatosis? Echocardiography. 2018;35(11):1772-81.
|
| 8. |
Lee PL, Beutler E, Rao S V, Barton JC. Genetic abnormalities and juvenile hemochromatosis: mutations of the HJV gene encoding hemojuvelin. Blood. 2004;103(12):4669-71.
|
| 9. |
Ravasi G, Rausa M, Pelucchi S, Arosio C, Greni F, Mariani R, et al. Transferrin receptor 2 mutations in patients with juvenile hemochromatosis phenotype. Am J Hematol. 2015;90(12):E226-7.
|
| 10. |
Charmandari E, Nicolaides NC, Chrousos GP. Adrenal insufficiency. Lancet. 2014;383(9935):2152-67.
|
| 11. |
Huang KE, Mittelman SD, Coates TD, Geffner ME, Wood JC. A significant proportion of thalassemia major patients have adrenal insufficiency detectable on provocative testing. J Pediatr Hematol Oncol. 2015;37(1):54-9.
|
| 12. |
Varkonyi J, Kaltwasser JP, Seidl C, Kollai G, Andrikovics H, Tordai A. Correspondence: A case of non-HFE Juvenile Haemochromatosis presenting with adrenocortical insufficiency. Br J Haematol. 2000;109(1):252-3.
|
| 13. |
Banaszkiewicz K, Sikorska K, Dorniak K, Lewczuk-Myślicka A, Sworczak K, Szurowska E, et al. Primary adrenal insufficiency and hemochromatosis — cause and effect relationship or a coincidence? Ann Endocrinol (Paris). 2018 Oct;[Epub ahead of print].
|
| 14. |
Uitz PM, Hartleb S, Schaefer S, Al-Fakhri N, Kann PH. Pituitary function in patients with hereditary haemochromatosis. Horm Metab Res. 2013;45(1):54-61.
|
| 15. |
McNeil LW, McKee Jr LC, Lorber D, Rabin D. The endocrine manifestations of hemochromatosis. Am J Med Sci. 1983;285(3):7-13.
|
| 16. |
Edwards CQ, Kelly TM, Ellwein G, Kushner JP. Thyroid disease in hemochromatosis: increased incidence in homozygous men. Arch Intern Med. 1983;143(10):1890-3.
|
| 17. |
Murphy MS, Walsh CH. Thyroid function in haemochromatosis. Ir J Med Sci. 2004;173(1):27-9.
|
| 18. |
Valenti L, Varenna M, Fracanzani AL, Rossi V, Fargion S, Sinigaglia L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int. 2009;20(4):549-55.
|
| 19. |
Guggenbuhl P, Deugnier Y, Boisdet JF, Rolland Y, Perdriger A, Pawlotsky Y, et al. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos Int. 2005;16(12):1809-14.
|
| 20. |
Messer JG, Kilbarger AK, Erikson KM, Kipp DE. Iron overload alters iron-regulatory genes and proteins, down-regulates osteoblastic phenotype, and is associated with apoptosis in fetal rat calvaria cultures. Bone. 2009;45(5):972-9.
|
| 21. |
Yamasaki K, Hagiwara H. Excess iron inhibits osteoblast metabolism. Toxicol Lett. 2009;191(2–3):211-5.
|
| 22. |
Doyard M, Chappard D, Leroyer P, Roth M-P, Loréal O, Guggenbuhl P. Decreased bone formation explains osteoporosis in a genetic mouse model of hemochromatosiss. PLoS One. 2016;11(2):e0148292.
|
| 23. |
O’Sullivan EP, McDermott JH, Murphy MS, Sen S, Walsh CH. Declining prevalence of diabetes mellitus in hereditary haemochromatosis — the result of earlier diagnosis. Diabetes Res Clin Pract. 2008;81(3):316-20.
|
| 24. |
Dubois-Laforgue D, Larger E, Timsit J. Is diabetes mellitus a sufficient condition to suspect hemochromatosis? Diabetes Metab. 2000;26(4):318-21.
|
| 25. |
Acton RT, Barton JC, Passmore L V, Adams PC, Speechley MR, Dawkins FW, et al. Relationships of serum ferritin, transferrin saturation, and HFE mutations and self-reported diabetes in the Hemochromatosis and Iron Overload Screening (HEIRS) study. Diabetes Care. 2006;29(9):2084-9.
|
| 26. |
Moczulski DK, Grzeszczak W, Gawlik B. Role of hemochromatosis C282Y and H63D mutations in HFE gene in development of type 2 diabetes and diabetic nephropathy. Diabetes Care. 2001;24(7):1187-91.
|
| 27. |
Davis TME, Beilby J, Davis WA, Olynyk JK, Jeffrey GP, Rossi E, et al. Prevalence, Characteristics, and Prognostic Significance of HFE Gene Mutations in Type 2 Diabetes: The Fremantle Diabetes Study. Diabetes Care. 2008 Sep 1;31(9):1795-801.
|
| 28. |
Rong Y, Bao W, Rong S, Fang M, Wang D, Yao P, et al. Hemochromatosis gene (HFE) polymorphisms and risk of type 2 diabetes mellitus: a meta-analysis. Am J Epidemiol. 2012;176(6):461-72.
|
