
Vascular complications in patients with Autosomal Dominant Polycystic Kidney Disease. A review of the literature and current clinical recommendations


Abstract
Autosomal dominant polycystic kidney disease is the most common genetic cause of renal failure. Apart from kidney involvement, patients are at risk of extra-renal manifestations, including vascular lesions. The etiology of vascular changes is diverse and depends, among other factors, on polycystin gene mutation, increased activity of the renin-angiotensin-aldosterone system and the occurrence of hypertension. The observed vascular system complications include cerebral artery aneurysms, cervico-encephalic arteries' dissection, aortic aneurysm and dissection and intracranial arterial dolichoectasia. This article discusses the etiopathogenesis, symptomatology, principles of prevention and treatment of the aforementioned diseases of the vascular system accompanying polycystic kidney disease.
Citation
Koska-Ścigała A, Zdrojewski Ł, Jankowska M, Dębska-Ślizień M A. Vascular complications in patients with Autosomal Dominant Polycystic Kidney Disease. A review of the literature and current clinical recommendations. Eur J Transl Clin Med. 2020;3(2):64-71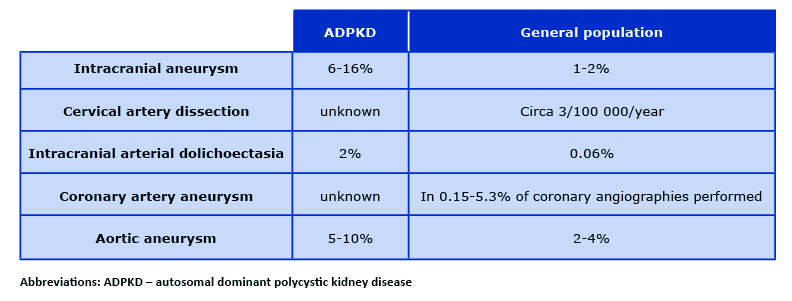


Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic cause of renal failure. Its incidence in the general population is estimated at 1/400-1/1000 people. ADPKD is a disease inherited in an autosomal dominant manner with an estimated 50% risk of transmission to offspring. It is estimated that approximately 10% of all patients requiring kidney replacement therapy are suffering from this disease. ADPKD is a systemic disorder which results in cystic changes in the kidneys, liver, pancreas, seminal vesicles, ovaries and central nervous system (CNS), as well as non-cystic complications such as inguinal hernia or diverticulitis. Among people with genetic mutations that cause ADPKD, changes in the cardiovascular system are also more frequently observed [1-2].
Material and methods
This is a narrative type of review. We conducted a search of Medline-Ovid database (1990 to December 2019) using a key: polycystic kidney disease and its abbreviations (e.g. PKD, ADPKD) combined with a term vascular. In the next step, we excluded all articles that did not reference ADPKD. The relevance and eligibility of the retrieved records were considered by two authors, independently.
Pathogenesis of vascular changes in ADPKD
Mutations in the genes encoding polycystin 1 (PKD1) and polycystin 2 (PKD2) are present in ADPKD. In individual cases, the disease may arise as a result of mutations also in other genes (e.g. GANAB and DNAJB11) [3]. The PKD-1 mutation occurs in about 85-90%, and PKD-2 in about 10-15% of people with a clinical manifestation of ADPKD. Both genes code for membrane proteins that are among the building blocks of fast calcium channels, responsible for intracellular transport. Disfunction of this channel leads to increase in cAMP and promotes cell proliferation. Synthesis of cAMP is also stimulated by vasopressin, whose concentration is higher in ADPKD patients compared to the general population [1-2, 4]. Both polycystin 1 and polycystin 2 are found in smooth muscle and vascular endothelium, therefore mutations in genes coding these proteins are responsible for damage to the vascular wall [5].
In ADPKD, endothelium damage occurs in the early stages of the disease. This is due to the reduced activity of nitric oxide synthase in the vascular endothelium, which results in impaired vasodilatation. Another vasoconstrictive factor in patients with ADPKD, is the elevated level of endothelin I. Endothelin I is produced by the epithelium of the renal cyst and secreted into its lumen [6-7]. In this way, the balance between vasodilatation and vasoconstriction is disturbed. In addition, renal cysts press on renal vessels which leads to ischemia of renal parenchyma and activation of the renin-angiotensin-aldosterone (RAA) system.
In the early stage of the disease, RAA stimulation leads to hypertension, which directly damages vascular system, and is the main symptom of ADPKD [6]. Hypertension is observed in 60% of adults with ADPKD with normal renal function [7]. The average age upon diagnosis of hypertension in these patients is 32 years for men and 34 years for women [8]. In addition, hypertension affects approximately 20-30% of children with ADPKD [6].
Atherosclerosis is more common in the course of ADPKD. Thickening of the inner and middle layer of the vessel allows to determine the degree of atherosclerosis. The intima media thickness (IMT) is a predictor of the development of cardiovascular diseases as well as an early marker of vascular complications. It was proposed that the IMT is greater in young patients and that the thickening of IMT occurs early in asymptomatic patients [9-10]. IMT is examined by Doppler ultrasound of the cervical arteries by measuring the thickness of the middle and inner membrane layers, 1 cm proximal to the division of the common carotid artery. In ADPKD patients with normal blood pressure and with normal kidney function, the IMT value is significantly higher than in the general population. This is crucial because cardiovascular diseases are the leading cause of death among ADPKD patients [9].
Intracranial aneurysms
Due to significant risk of death and disability in the course of CNS bleeding, intracranial aneurysms (also called cerebral or brain aneurysms) are the most dangerous of all the extra-renal manifestations of ADPKD [11]. They occur in about 6% of patients with ADPKD without any family history of intracranial aneurysms and as many as 16% of patients with such family history [12]. In the general population, the incidence of intracranial aneurysms is estimated at 1-2% [13] (Table 1). Their occurrence is slightly more common in women, the incidence increases with age, and the peak of their occurrence, according to different authors, falls on the fourth or sixth decade of life [14-16]. There are two types of intracranial aneurysms (85% are saccular and 15% are fusiform) and they form as a result of damage to the elastic central membrane of the vessel [17]. 90% of the aneurysms are located in the anterior circulation of the brain, most often in the internal carotid, middle cerebral and anterior communicating arteries). Posterior circulation aneurysms are much less frequent. Most of the aneurysms have a diameter < 6mm, with an average of 3.5 mm (3 mm for women and 4 mm for men) [16-18].
Table 1. The incidence of vascular complications in patients with ADPKD
In most cases, intracranial aneurysms grow asymptomatically. Some of the aneurysms cause a local mass effect, sometimes the cerebral ischemia is caused by clot material released from the dome of the aneurysm. However, an aneurysm rupture and SAH (subarachnoid hemorrhage) is observed in only about 0.3-2.3% of the cases [16]. The risk of aneurysm rupture increases with its diameter. Depending on their location, aneurysms located in the posterior circulation rupture more often. In the case of anterior circulation, the risk of hemorrhage increases when the diameter of the aneurysm is > 7 mm. In the posterior cerebral circulation, it is independent of the diameter [19]. Additional independent risk factors for aneurysm rapture and SAH are alcohol abuse, nicotinism and poor blood pressure control [20].
The main symptom of aneurysm rupture is sudden and severe headache (often referred to as "the most intense headache of my life"). It is also worth to remember about the so-called warning pains that often occur few weeks earlier. About half of the patients lose consciousness. Epileptic seizures appear in approximately 10% of patients with subarachnoid hemorrhage. Meningeal signs are not specific for SAH. We observe neurological symptoms depending on location and extent of the haemorrhage [17]. The diagnostic method of choice in cerebral hemorrhage is non-contrast computed tomography (CT). In the absence of blood in CT scans, a lumbar puncture should be performed to confirm the presence of erythrocytes or haemolysis products (bilirubin, oxyhemoglobin) in the cerebrospinal fluid [17].
There are two available methods for the treatment of aneurysms: neurosurgical clipping and endovascular. The first method requires craniotomy, therefore it carries a surgery-related risk for the patient, e.g. clip slippage due to incomplete clipping and insufficient amount of used clips [17]. Vascular clips are placed on aneurysms which are not appropriate for endovascular treatment, e.g. large aneurysms and those with a wide neck. The endovascular method is based on embolization of the aneurysm (by means of so-called coils) and excluding it from the cerebral circulation. This treatment is less risky and reduces the danger of patient's disability. In case of SAH, endovascular embolization should be performed up to 3 days from the onset of symptoms [12, 17].
Preventive treatment consists of excluding aneurysms with potential for rupture from the circulation and has significant challenges. On the one hand, this procedure has the potential to significantly reduce the mortality of patients with cerebral artery aneurysm and ADPKD. On the other hand, it is necessary to take into account the increased surgery risk, frequent detection of small aneurysms (with low risk of rupture) in brain imaging. In addition, at the moment there is a lack of reliable radiological, biochemical or genetic markers that would help assess the risk of aneurysm rupture in an ADPKD patient, thus justifying a preventive procedure [22].
The most common complication of bleeding from a ruptured aneurysm is re-bleeding and secondary CNS ischemia. About half the of patients with SAH die and 10-20% of those who survive remain severely disabled. There is also an increased incidence of another rupture in the cerebral artery aneurysm in patients who have had such an episode in the past [17].
Monitoring of intracranial aneurysms in patients with ADPKD
The method of choice in the imaging of intracerebral aneurysms is magnetic resonance angiogram (MRA). This method does not require use of any contrast agents, thus eliminating the risk of nephrotoxicity (as opposed to angio-CT) or systemic complications (as in the case of gadolinium-based contrast in MR) in patients with impaired renal function. Its limitations are contraindications to perform MR, such as the presence of metal implants, vascular clips or lack of patient’s cooperation with the procedure [23].
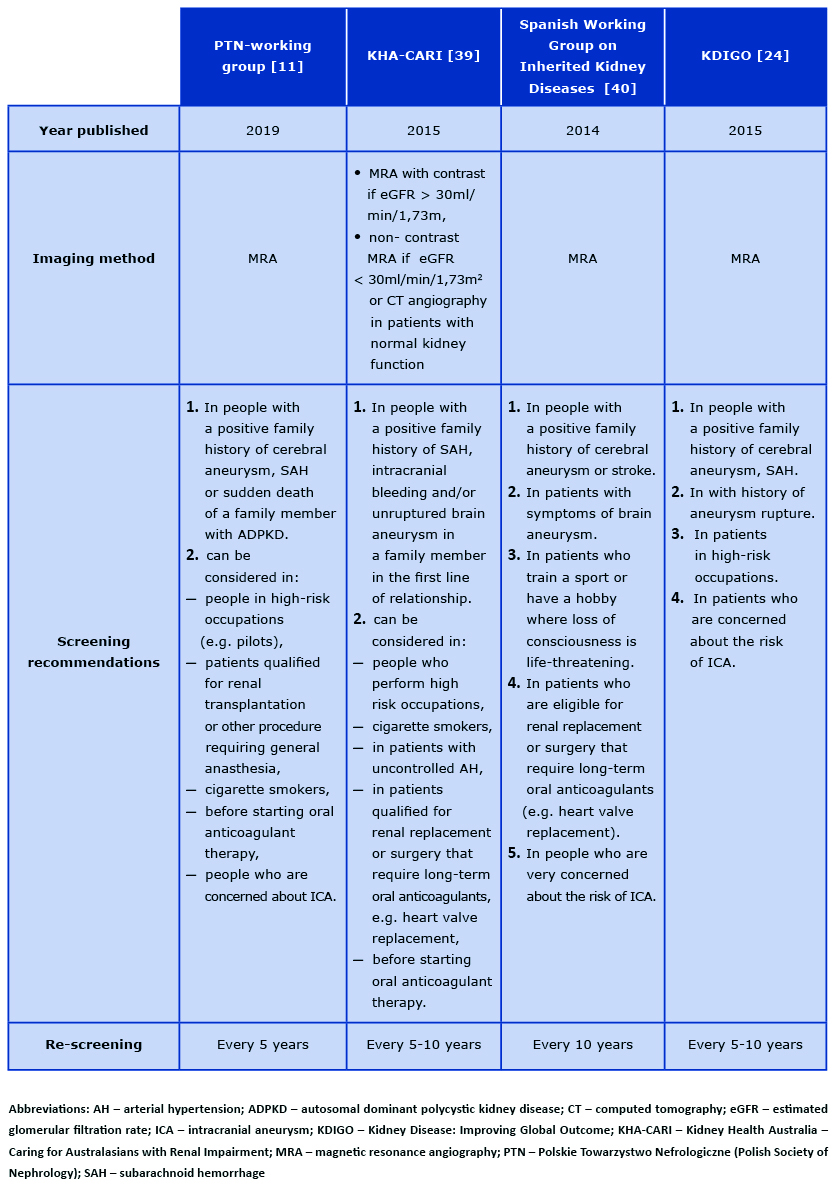
The indication for screening via MRA of the brain is the presence of symptoms suggesting an aneurysm or family history of cerebral aneurysm or intracranial hemorrhage. This imaging should also be performed in the group of ADPKD patients performing high-risk occupations, e.g. pilots, drivers [15]. In this group of patients, the screen should be repeated every 5 years. In patients with a family history of aneurysms, MRA should be performed every 5-10 years [13, 24-26]. Recent literature suggests the benefits of conducting screening tests also in patients without any family history, i.e. in cigarette smokers, in patients before starting oral anticoagulants, in patients qualified for renal transplantation or surgery requiring long-term oral anticoagulants and in those who are very concerned about the risk of brain aneurysm [27] (see Table 2).
Table 2. An overview of the recommendations for monitoring intracranial aneurysms in patients with ADPKD
Cervico-encephalic artery dissection
Cervico-encephalic artery dissection is a relatively rare complication of ADPKD. Its incidence in the general population is estimated at 3/1000 people, however specific data about incidence in the ADPKD population is not available [16]. The most frequently affected vessels are the internal carotid artery (~75% of cases) and vertebral artery (~15%). We distinguish two types of dissection: spontaneous and post-traumatic. The etiological factors causing artery dissection in ADPKD include arterial hypertension and defects in the vessel wall structure. Dissection occurs as a result of damage to the inner membrane, its separation and the passage of blood between the inner and middle membrane of the vessel, thus creating the so-called false lumen. Post- -traumatic dissection usually occurs in younger patients (about 30 years of age), in contrast to spontaneous dissection whose peak is observed about 50 years of age.
The main symptom of internal carotid artery dissection is a severe headache, most often in the frontotemporal area. The pain is usually develops gradually, is not sudden and is accompanied by symptoms of focal neurological signs and Horner syndrome. In the case of dissection of the vertebral artery, the pain appears in the occipital region, in addition to neurological signs from the vertebrobasilar circulation.
Doppler ultrasound examination is the method of choice in case of suspected cervico-encephalic dissection, which should be confirmed with MR (FAT-SAT sequence) with cerebral arteries assessment. Treatment consists of taking oral anticoagulants for 3-6 months [28-30].
Intracranial arterial dolichoectasia
Dolichoectasia is the elongation and widening of the vessels. Specifically, the damage occurs to the inner and middle membrane of the vessel. In the brain, dolichoectasia usually occurs in the vertebral arteries and the basilar artery. This pathology occurs in approximately 2% of patients with ADPKD, compared to 0.06% in the general population [30]. Most patients with dolichoectasia are asymptomatic and this change is detected accidentally. Rarely, patients suffer ischemic stroke in the course of basal/vertebral artery clot or embolism caused by thrombotic material. Occasionally, cranial nerves are damaged, increased intracranial pressure and hydrocephalus take place. Surgical treatment is not recommended in the case of intracranial artery dolichoectasia [31].
Aneurysms of coronary arteries
In addition to classic ADPKD complications in the myocardium, such as hypertrophy or valve defects (the most common is mitral valve prolapse), cases of coronary aneurysms are described [32]. The criterion for the diagnosis of coronary aneurysm is > 1.5-fold widening of the vascular lumen in relation to the diameter of the widest coronary vessel in the patient. Right coronary artery and anterior descending artery aneurysms are the most common, whereas circumflex artery and left coronary artery aneurysms are less frequent. Coronary aneurysms may be asymptomatic. However, angina pain often occurs due to myocardial ischemia secondary to embolic material released from the aneurysm lumen. The complication may also be acute coronary syndrome and arrhythmias. Very rarely, the ruptured aneurysm may cause pericardial tamponade. Small aneurysms do not usually require invasive treatment such as endovascular stent implantation and cardiac surgery (in the case of multiple aneurysms or those of very large sizes) [33].
Aortic aneurysm
Aortic aneurysms in patients with ADPKD are most often found in the abdominal part, mainly in patients with advanced renal failure requiring dialysis. The incidence is estimated at approximately 5-10% of patients, compared to 2-4% in the general population. It is controversial whether or not the formation of aortic aneurysms is directly related to kidney disease or results from hypertension and atherosclerotic lesions that are common in patients with ADPKD. Currently there are no indications to screen for aortic aneurysms in patients with ADPKD differently than in the general population [13]. In Poland, as a part of the National Screening Program for Abdominal Aorta aneurysm in 2018-2020, people over 65 years of age who have at least 3 of the risk factors (e.g. hypertension, male gender, hyperlipidemia, coronary disease and tobacco smoking) are eligible for screening. Similar screening programs are offered in other countries, e.g. the United Kingdom. The possibility of aortic dissection or rupture of the abdominal aortic aneurysm should be considered in every patient with ADPKD, who experienced sudden severe abdominal pain of unknown origin. For technical reasons, visualising abdominal aorta aneurysms using ultrasound may be hindered by the enlarged polycystic kidneys.
Thoracic aortic dissections are relatively rare in patients with ADPKD, while they are characteristic of patients with Marfan syndrome. Two types of dissection are distinguished according to the Stanford classification: type A (the dissection includes the ascending aorta with or without the descending part) and type B (dissection occurs only in the descending aorta). Type A dissection is noted in about 60-70% of patients, whereas the rest have type B dissection. In type A dissection the mortality is about 50% and these patients require surgical treatment. Severe chest pain radiating to the back in all patients is a life-threatening condition and may mean dissection of the aortic thoracic segment [34-36].
Future perspectives
An increasing number of studies provide recommendations for monitoring aneurysm in ADPKD, however there are some points not clearly defined, such as the patient’s age at screening, the frequency and duration of repeat screening, the use of follow-up imaging after intracranial aneurysm diagnosis. Recent literature suggests that screening for intracranial aneurysms with MR angiography in all patients with autosomal dominant polycystic kidney disease is cost-effective [37]. The high prevelance of aortic aneurysm is estimated in patients with ADPKD. Larger studies are needed to confirm the utility of specific echocardiographic screening as currently there is no such recommendation [38].
Conclusions
Polycystic kidney disease inherited in an autosomal dominant way is a disorder with many organs involvement. In some patients, non-renal symptoms, including vascular changes, occur. Mutations in the polycystin 1 and 2 gene are considered the main cause of vasculopathy. In the course of vascular wall damage, brain aneurysms may occur, the rupture of which leads to intracranial hemorrhage and often significant neurological deficits. It is very important to monitor patients from so-called risk groups for intracranial aneurysms – this allows the detection and treatment of aneurysms before they rupture. Cervico-encephalic dissections, aortic aneurysm and dissection, and cerebral vascular dolichoectasia are more common in ADPKD than in the general population. It has been proven that atheromatic changes happen much faster in patients with ADPKD and increase the risk of cardiovascular diseases, which are the main cause of death in these patients. The proper prophylaxis and reduction of vascular risk factors through correct treatment of hypertension, lipid disorders, cessation of smoking and alcohol abuse, as well as, propagation of other pro-health behaviors are of key importance for slowing down disease progression and reducing mortality.
Conflict of interest: none declared
References
| 1. |
Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet [Internet]. 2007;369(9569):1287–301. Available from: https://doi.org/10.1016/S0140-6736(07)60601-1.
|
| 2. |
Chow CL, Ong ACM. Autosomal dominant polycystic kidney disease. Clin Med [Internet]. 2009 Jun;9(3):278–83. Available from: https://pubmed.ncbi.nlm.nih.gov/19634398.
|
| 3. |
Lipska-Ziętkiewicz B, Klinger M, Różański J, Nowicki M, Augustyniak-Bartosik H, Szurowska E, et al. Rekomendacje Grupy Roboczej PTN. Zasady postępowania z chorymi na autosomalną dominującą wielotorbielowatość nerek i inne torbielowate choroby nerek: Diagnostyka molekularna i poradnictwo genetyczne w ADPKD. Nefrol i Dializoterapia Pol [Internet]. 2018;22:91–3. Available from: https://ptnefro.pl/index.php/content/download/4717/67698/file/Rekomendacje Grupy Roboczej PTN.pdf.
|
| 4. |
Ong ACM, Harris PC. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int [Internet]. 2005;67(4):1234–47. Available from: https://doi.org/10.1111/j.1523-1755.2005.00201.x.
|
| 5. |
Kang YR, Ahn J-H, Kim KH, Choi YM, Choi J, Park JR. Multiple Cardiovascular Manifestations in a Patient with Autosomal Dominant Polycystic Kidney Disease. J Cardiovasc Ultrasound [Internet]. 2014 Sep 29;22(3):144–7. Available from: http://dx.doi.org/10.4250/jcu.2014.22.3.144.
|
| 6. |
Ecder T, Schrier RW. Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat Rev Nephrol [Internet]. 2009;5(4):221–8. Available from: https://doi.org/10.1038/nrneph.2009.13.
|
| 7. |
Ecder T, Schrier RW. Hypertension in Autosomal-Dominant Polycystic Kidney Disease: Early Occurrence and Unique Aspects. J Am Soc Nephrol [Internet]. 2001 Jan 1;12(1):194 LP – 200. Available from: http://jasn.asnjournals.org/content/12/1/194.abstract.
|
| 8. |
Schrier RW, Johnson AM, Mcfann K, Chapman AB. The role of parental hypertension in the frequency and age of diagnosis of hypertension in offspring with autosomal-dominant polycystic kidney disease. Kidney Int [Internet]. 2003 Nov;64(5):1792–9. Available from: http://www.sciencedirect.com/science/article/pii/S0085253815495316.
|
| 9. |
Kocaman O, Oflaz H, Yekeler E, Dursun M, Erdogan D, Demirel S, et al. Endothelial dysfunction and increased carotid intima-media thickness in patients with autosomal dominant polycystic kidney disease. Am J Kidney Dis [Internet]. 2004;43(5):854–60. Available from: http://www.sciencedirect.com/science/article/pii/S0272638604001386.
|
| 10. |
Ecder T. Cardiovascular complications in autosomal dominant polycystic kidney disease. Curr Hypertens Rev [Internet]. 2013;9(1):2–11. Available from: https://www.ingentaconnect.com/content/ben/chyr/2013/00000009/00000001/art00002.
|
| 11. |
Dębska-Ślizień, Alicja Jankowska M, Nowicki M, Klinger M, Augustyniak-Bartosik H, Limon J, Lipska-Ziętkiewicz B. Grupa Robocza Polskiego Towarzystwa Nefrologicznego – Zasady postępowania z chorymi na autosomalnie dominujące wielotorbielowate zwyrodnienie nerek (ADPKD) i inne torbielowate choroby nerek. Nefrol i Dializoterapia Pol [Internet]. 2019;23(1):1–15. Available from: http://docplayer.pl/171553352-Diagnostyka-adpkd-adpkd-jest-choroba-dziedziczona-w-sposob-autosomalny-dominujacy-w-typowych.html.
|
| 12. |
Pirson Y, Chauveau D, Torres V. Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol [Internet]. 2002;13(1):269–76. Available from: https://jasn.asnjournals.org/content/13/1/269.short.
|
| 13. |
Luciano RL, Dahl NK. Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol Dial Transplant [Internet]. 2014 Feb 1;29(2):247–54. Available from: https://doi.org/10.1093/ndt/gft437.
|
| 14. |
H.W. X, Qiang YS, Lin MC, Hua LM. Screening for Intracranial Aneurysm in 355 Patients With Autosomal-Dominant Polycystic Kidney Disease. Stroke [Internet]. 2011 Jan 1;42(1):204–6. Available from: https://doi.org/10.1161/STROKEAHA.110.578740.
|
| 15. |
Schrier RW, Belz MM, Johnson AM, Kaehny WD, Hughes RL, Rubinstein D, et al. Repeat Imaging for Intracranial Aneurysms in Patients with Autosomal Dominant Polycystic Kidney Disease with Initially Negative Studies: A Prospective Ten-Year Follow-up. J Am Soc Nephrol [Internet]. 2004 Apr 1;15(4):1023 LP – 1028. Available from: http://jasn.asnjournals.org/content/15/4/1023.abstract.
|
| 16. |
Irazabal M V, Huston J, Kubly V, Rossetti S, Sundsbak JL, Hogan MC, et al. Extended follow-up of unruptured intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol [Internet]. 2011;6(6):1274–85. Available from: https://cjasn.asnjournals.org/content/6/6/1274.short.
|
| 17. |
Kobayashi A, Członkowska A. Neurologia. Medical Tribune Polska; 2014. 230–267 p.
|
| 18. |
Belz MM, Hughes RL, Kaehny WD, Johnson AM, Fick-Brosnahan GM, Earnest MP, et al. Familial clustering of ruptured intracranial aneurysms in autosomal dominant polycystic kidney disease. Am J Kidney Dis [Internet]. 2001;38(4):770–6. Available from: http://www.sciencedirect.com/science/article/pii/S027263860163717X.
|
| 19. |
Wiebers DO. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet [Internet]. 2003;362(9378):103–10. Available from: http://www.sciencedirect.com/science/article/pii/S0140673603138603.
|
| 20. |
Różański J. Aktualny stan wiedzy na temat ADPKD. In: Forum Nefrologiczne [Internet]. 2012. p. 140–7. Available from: https://journals.viamedica.pl/forum_nefrologiczne/article/download/19176/15094.
|
| 21. |
Szmuda T, Słoniewski P. Late postoperative slippage of the cerebral aneurysm clip. A systematic review and meta-analysis. Eur J Transl Clin Med [Internet]. 2019 Jun 7;2(1):56–69. Available from: https://doi.org/10.31373/ejtcm/103442.
|
| 22. |
Niemczyk M. Treatment of unruptured intracranial aneurysms in autosomal dominant polycystic kidney disease: primum non nocere. Am J Neuroradiol [Internet]. 2016;37(2):294–5. Available from: https://doi.org/10.3174/ajnr.A4538.
|
| 23. |
Schievink WI, Torres VE, Piepgras DG, Wiebers DO. Saccular intracranial aneurysms in autosomal dominant polycystic kidney disease. J Am Soc Nephrol [Internet]. 1992;3(1):88–95. Available from: https://jasn.asnjournals.org/content/3/1/88.short.
|
| 24. |
Chapman AB, Devuyst O, Eckardt K-U, Gansevoort RT, Harris T, Horie S, et al. Autosomal-dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int [Internet]. 2015;88(1):17–27. Available from: https://doi.org/10.1038/ki.2015.59.
|
| 25. |
Zhou Z, Xu Y, Delcourt C, Shan J, Li Q, Xu J, et al. Is regular screening for intracranial aneurysm necessary in patients with autosomal dominant polycystic kidney disease? A systematic review and meta-analysis. Cerebrovasc Dis [Internet]. 2017;44(1–2):75–82. Available from: https://www.karger.com/Article/Abstract/476073.
|
| 26. |
Niemczyk M, Gradzik M, Fliszkiewicz M, Kulesza A, Gołębiowski M, Pączek L. Natural history of intracranial aneurysms in autosomal dominant polycystic kidney disease. Neurol Neurochir Pol [Internet]. 2017;51(6):476–80. Available from: https://www.sciencedirect.com/science/article/pii/S0028384317302529.
|
| 27. |
Flahault A, Trystram D, Nataf F, Fouchard M, Knebelmann B, Grünfeld J-P, et al. Screening for intracranial aneurysms in autosomal dominant polycystic kidney disease is cost-effective. Kidney Int [Internet]. 2018;93(3):716–26. Available from: http://www.sciencedirect.com/science/article/pii/S0085253817306130.
|
| 28. |
Schievink WI. Spontaneous Dissection of the Carotid and Vertebral Arteries. N Engl J Med [Internet]. 2001 Mar 22;344(12):898–906. Available from: https://doi.org/10.1056/NEJM200103223441206.
|
| 29. |
Misztal M, Kwiatkowska W, Ohly P, Nessler J. Internal carotid artery dissection--symptomatology, diagnosis and treatment. Kardiol Pol [Internet]. 2011;69(9):958–62. Available from: https://ruj.uj.edu.pl/xmlui/bitstream/handle/item/244012/nessler_et-al_rozwarstwienie_tetnicy_szyjnej_wewnetrznej_2011.pdf?sequence=1&isAllowed=y.
|
| 30. |
Kuroki T, Yamashiro K, Tanaka R, Hirano K, Shimada Y, Hattori N. Vertebral Artery Dissection in Patients with Autosomal Dominant Polycystic Kidney Disease. J Stroke Cerebrovasc Dis [Internet]. 2014;23(10):e441–3. Available from: http://www.sciencedirect.com/science/article/pii/S1052305714002730.
|
| 31. |
Schievink WI, Torres VE, Wiebers DO, Huston J. Intracranial arterial dolichoectasia in autosomal dominant polycystic kidney disease. J Am Soc Nephrol [Internet]. 1997;8(8):1298–303. Available from: https://jasn.asnjournals.org/content/8/8/1298.short.
|
| 32. |
Pirson Y. Extrarenal Manifestations of Autosomal Dominant Polycystic Kidney Disease. Adv Chronic Kidney Dis [Internet]. 2010;17(2):173–80. Available from: http://www.sciencedirect.com/science/article/pii/S1548559510000042.
|
| 33. |
Araszkiewicz A, Grygier M, Lesiak M, Grajek S. From positive remodelling to coronary artery ectasia. Is coronary artery aneurysm a benign form of coronary disease? Kardiol Pol. 2009;67(12):1390.
|
| 34. |
Zagrodzka M, Domaradzki W, Young E. Vademecum radiologiczne kardiologa i kardiochirurga – rozwarstwienie aorty piersiowej. Kardiol po Dyplomie [Internet]. 2011;10(3):86–100. Available from: https://podyplomie.pl/publish/system/articles/pdfarticles/000/009/044/original/86-100.pdf?1468921842.
|
| 35. |
Adeola T, Adeleye O, Potts JL, Faulkner M, Oso A. Thoracic aortic dissection in a patient with autosomal dominant polycystic kidney disease. J Natl Med Assoc [Internet]. 2001;93(7–8):282. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2594041/.
|
| 36. |
Dimarakis I, Kadir I. Acute aortic syndrome in autosomal dominant polycystic kidney disease. Kidney Int [Internet]. 2017 Feb;91(2):512. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0085253816305464.
|
| 37. |
Malhotra A, Wu X, Matouk CC, Forman HP, Gandhi D, Sanelli P. MR Angiography Screening and Surveillance for Intracranial Aneurysms in Autosomal Dominant Polycystic Kidney Disease: A Cost-effectiveness Analysis. Radiology [Internet]. 2019 May;291(2):400–8. Available from: http://pubs.rsna.org/doi/10.1148/radiol.2019181399.
|
| 38. |
Bouleti C, Flamant M, Escoubet B, Arnoult F, Milleron O, Vidal-Petiot E, et al. Risk of Ascending Aortic Aneurysm in Patients With Autosomal Dominant Polycystic Kidney Disease. Am J Cardiol [Internet]. 2019;123(3):482–8. Available from: http://www.sciencedirect.com/science/article/pii/S0002914918320459.
|
| 39. |
Rangan GK, Lee VW, Alexander SI, Patel C, Tunnicliffe DJ, Vladica P. KHA-CARI Autosomal Dominant Polycystic Kidney Disease Guideline: Screening for Polycystic Kidney Disease. Semin Nephrol [Internet]. 2015 Nov;35(6):557-564.e6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0270929515001680.
|
| 40. |
Ars E, Bernis C, Fraga G, Martínez V, Martins J, Ortiz A, et al. Spanish guidelines for the management of autosomal dominant polycystic kidney disease *. Nephrol Dial Transplant [Internet]. 2014 Sep 1;29(suppl_4):iv95–105. Available from: https://doi.org/10.1093/ndt/gfu186.
|
