

Game changer: understanding and managing hypertrophic cardiomyopathy in athletes


Abstract
Physiological left ventricular hypertrophy in athletes often blurs the diagnostic line with hypertrophic cardiomyopathy (HCM), necessitating a critical differentiation to avert sudden cardiac death (SCD). This review explores various diagnostic approaches and proposes a well-founded strategy. The electrocardiogram (ECG) emerges as a highly sensitive HCM diagnostic tool, revealing characteristic changes in over 90% of cases. Holter monitoring aids in arrhythmia detection, yet its role in estimating SCD risk is subject to debate. Echocardiography encounters challenges in distinguishing HCM from normal athlete hearts. Cardiac magnetic resonance imaging (CMRI) enhances diagnostics by uncovering focal hypertrophy and fibrosis, with the absence of these markers not excluding HCM. Stress tests and family history provide invaluable diagnostic clues, and while genetic testing is not routine, its potential in uncovering hereditary factors is promising. Mandatory limitations on athletic activity for those with HCM are justified, given the heightened SCD risk during high-intensity sports. Extended diagnostic tests in borderline cases and universal screening for athletes are imperative for accurate risk assessment and the implementation of preventive measures. This comprehensive strategy integrates diverse diagnostic tools, ensuring a timely and precise identification of HCM, thereby mitigating the risk of SCD in the athletic community.
Citation
Kulak M, Sokołowska K, Bereza D, Moreau I, Polańska P, Lang M, Woch B, Lange N. Game changer: understanding and managing hypertrophic cardiomyopathy in athletes. Eur J Transl Clin Med. 2024;7(2):100-110





Introduction
Hypertrophic cardiomyopathy (HCM) is the most prevalent form of primary cardiomyopathy, with an estimated incidence of 1 in 500 people, regardless of age [1]. The main characteristic of this disease is an increase of left ventricular wall thickness (LVWT), measured at end-diastole in any segment) reaching ≥ 15 mm. This hypertrophy cannot be convincingly explained by any other debilitating heart disease, (e.g, aortic stenosis, hypertension, ischemic heart disease) or endocrine disorders. With LVWT < 15 mm, HCM can be diagnosed if there are additional features of the disease, e.g. positive family history (particularly with unexplained LVWT > 13 mm in the first-degree relatives of the affected individual), myocardial fibrosis confirmed by cardiac magnetic resonance imaging (CMRI), asymmetric hypertrophy seen in echocardiography (echo) or abnormalities in the electrocardiogram (ECG) [2]. However, the presence of structural dysfunction (e.g. changes in papillary muscle structure) or the systolic anterior motion of the mitral valve (although its incidence reaches as high as 1/3 of HCM cases), is not mandatory to make the diagnosis of HCM [3-4]. Although HCM is inherited in an autosomal dominant manner and is linked to more than 1400 mutations in at least 11 major genes, pinpointing a specific mutation is not necessary for diagnosis [5].
Regular exercise, particularly endurance sports (e.g. cross-country skiing, rowing, long-distance cycling and running, swimming, soccer and tennis), can also cause myocardial hypertrophy and an increase in heart dimensions by about 10-20% as an expression of adaptation to a greater load [6-8]. It has been proven that the heart mass of athletes is greater than that of the population with a sedentary lifestyle [9]. Thus, a diagnostic problem of inestimable importance for athletes arises, because the diagnosis of HCM, which is not excluded by satisfactory physical performance [10], makes it necessary to significantly limit the physical activity undertaken even in the absence of symptoms and high-risk indicators, e.g. the presence of left ventricular outflow tract (LVOT) stenosis (either at rest or during physical exercise) or late gadolinium enhancement (LGE) in CMRI (indicating fibrosis) [11]. Our aim was to further dissect these diagnostic intricacies in order to contribute to the understanding and facilitating informed decision-making in the care of HCM in athletes.
Materials and methods
A comprehensive literature review was undertaken to gather insights into HCM among athletes and its distinction from the athlete’s heart. Utilizing databases such as PubMed and Google Scholar, relevant studies focusing on HCM in athletes were identified using search terms like “hypertrophic cardiomyopathy,” “diagnosis,” “sudden cardiac death,” “genetic mutations,” and “athlete’s heart” including their Polish equivalents. The search was limited to articles published in English and Polish. Articles were included if they examined specific aspects of hypertrophic cardiomyopathy (HCM) in athletes (e.g. its pathophysiology, clinical symptoms, diagnostic methods, treatments, genetic mutations and the latest developments). Studies that were not relevant to the above-mentioned topics (or were duplicated) were excluded.
Results
The initial search retrieved a pool of 100 abstracts. 65 articles comprising reviews, case reports, original research and clinical trials were meticulously selected based on their direct relevance to HCM in athletes, excluding duplicates or unrelated content.
Risk of sudden cardiac death
As mentioned in the Introduction, strict recommendations persist in the guidelines because when primary (and often undetected, asymptomatic) HCM is present, participating in high-intensity competitive sports can promote ventricular tachycardia/ventricular fibrillation. These arrhythmias consequently constitute a potent (yet modifiable) independent risk factor of sudden cardiac death (SCD), even if there are no traditional risk markers linked to the HCM [11]. This most likely occurs through the unpredictable exacerbation of electrophysiological abnormalities in HCM by stress, changes in blood volume, electrolyte concentrations and catecholamine output during sports [12-13]. Strongly in favor of restrictions for athletes with HCM is the fact that in 2007, established or suspected HCM accounted for 44% of SCD in young athletes [14]. In recent years, this percentage has decreased to 14.1% and normal heart structure was more common in SCD cases [15]. This is likely due to the implementation of screening tests (e.g. ECG, physical examination, blood pressure measurement) in mandatory sports medicine examinations, which have significantly aided in identifying athletes at risk of SCD [16]. SCD in young athletes is quite rare (estimated at 1:50,000 people), but it is noted that there are many predicted years of healthy life for those affected and the mortality rate is 2.8-fold higher, compared to the non-athlete, same age population. A considerable portion of these untimely deaths stem from hereditary, asymptomatic heart abnormalities that manifest during physical exertion [17-18]. Hence, the need for screening seems all the more justified in this group, and The European Society of Cardiology (ESC) advises formal examination for all young professional athletes (usually 12-14 years of age) including history-taking and physical examination, family history gathering, and 12-lead ECG [19-20]. However, it should be emphasized that over 90% of exercise-induced SCD cases occur in amateur athletes, in whom screening options are nonetheless limited [21]. Furthermore, the incidence of SCD in a 10-year observational study of 10.9 million marathoners and half-marathoners were much lower, reaching 1:259,000, with HCM remaining the most important cause, confirmed in post-mortem studies [22-23]. Furthermore, after identifying patients via initial tests, a highly efficient risk stratification algorithm (proposed by AHA and ESC) can be applied to implement primary prevention of SCD [2, 14]. This involves the implantation of a cardioverter-defibrillator (ICD), which has significantly lowered the HCM-related mortality to 0.5% per year in the general population [12]. Prophylactic ICD implantation is not advised for athletes who do not meet the general indications to allow them to participate in competitive sports due to possible device-related complications (e.g. lead displacement (3.1%), infection at the implantation site (1.5%), swelling, bleeding or bruising, haematoma (1.2%), pneumothorax (1.1%) [11].
Electrocardiography
Electrophysiological manifestations of training, on the ECG, are generally divided into those caused by increased parasympathetic activity and those resulting from altered dimensions of the heart cavities. Some healthy athletes may have bradycardia or sinus arrhythmia at rest (usually correlated with respiratory rhythm), nodal rhythm, 1st degree atrioventricular (AV) block or 2nd degree Mobitz 1 AV block, which resolves with mild exercise. As for ventricular conduction, the criteria for left and/or right ventricular hypertrophy, incomplete right bundle branch block and sometimes J-point elevation with ascending ST segments may be met [25-26]. The elevated ST segment typically has a concave shape in the posterior and lateral leads, while convex elevation in the precordial leads is infrequent [27]. In the context of athletes, even if there’s cardiac axis deviation and the criteria for atrial and/or ventricular enlargement are met on the ECG [28], we consider these as variants within the normal range [29]. Unless accompanied by symptoms (physical or subjective) and family history, there’s usually no need for further diagnostic investigation. As with structural changes of the heart, male endurance athletes present the most ECG changes. In athletes younger than 14 years of age, there are differences in the changes presented, with more frequent T-wave inversion in the V1-V4 leads, although after 16 years of age these changes revolve spontaneously [30]. Therefore, in all Caucasian athletes ≥ 16 years of age, T-wave inversions in leads other than III, aVL, aVR, or V1 should raise diagnostic concerns [27, 31].
At the same time, ECG stands out as the most sensitive test for HCM, showing abnormalities in over 90% of cases [32]. Characteristic and most common abnormalities in HCM not seen in healthy athletes are deep T-wave inversions in lateral and/or posterior leads (occurring in 93% regardless of the amount of physical activity), horizontal or downsloping ST segment depression (in 47%). Other less common include pathological Q-waves (Q/R ratio > 0.25), in 27% and left bundle branch block. On the other hand, deep inverted T-waves (so-called “giant negative T waves”) in the precordial leads may be indicative of the mid-ventricular or apex variant of HCM [26, 29, 33-34]. The aforementioned changes, even despite the absence of abnormalities in echo, should prompt greater diagnostic vigilance and broader investigations (see Table 1).
Table 1. The most common electrocardiogram (ECG) changes in HCM|
Dynamic Holter ECG
Holter ECG is not strictly a diagnostic tool for HCM, but it is often essential for capturing co-occurring arrhythmias. Recent studies have demonstrated the superiority of 14-day monitoring over the routinely used 24-48-hour monitoring in detecting non-sustained ventricular tachyarrhythmias (NSVTs, defined as lasting < 30 seconds) which are the most prevalent cause of SCD in HCM. Multiple bursts of NSVT are associated with increased risk (particularly in young patients) and should encourage further diagnostics. Another possible arrhythmia is ventricular tachycardia (VT), which if intolerable for the patient could be an indication for either implanting an ICD or treatment with β-blockers or amiodarone for secondary prevention. In other cases, starting treatment is not advised, as there is no evidence to suggest that sustained (> 30 seconds), monomorphic and well-tolerated VT predisposes to SCD more than non-sustained ventricular tachycardia (NSVT) [2, 35].
Echocardiography
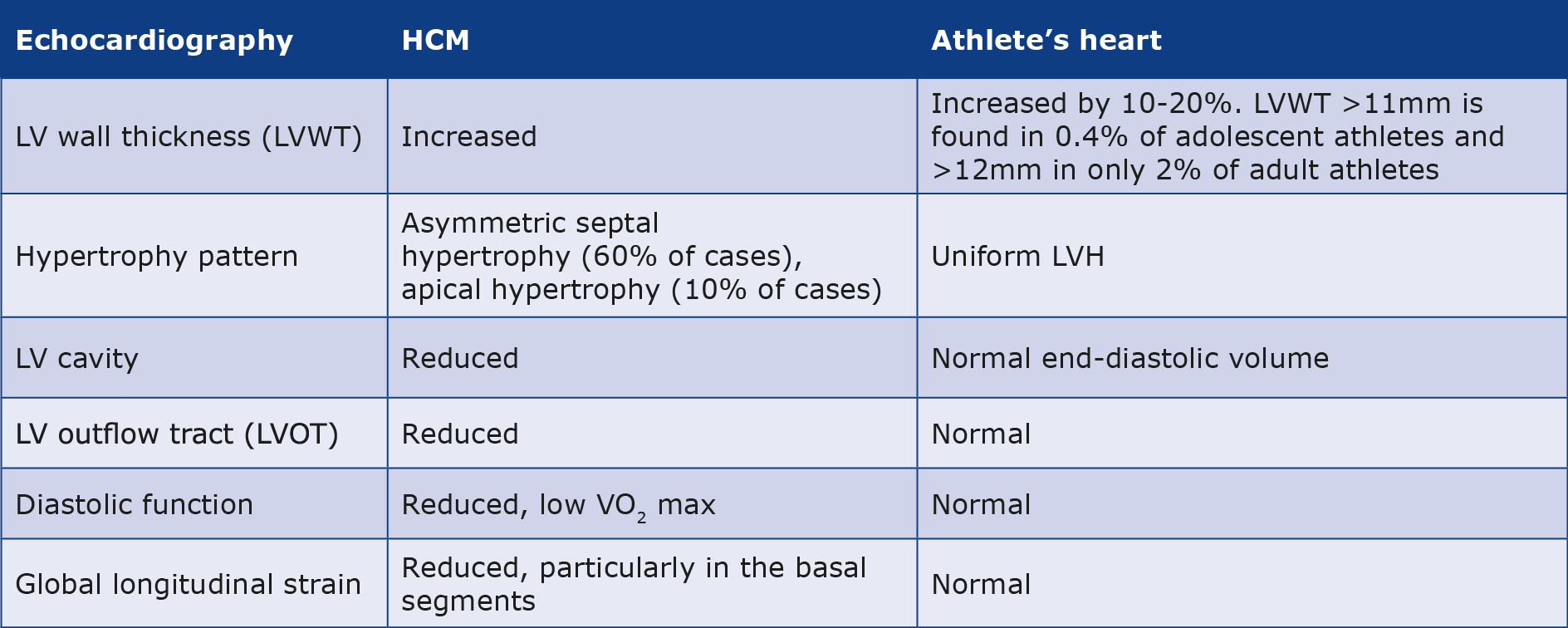
Frequently observed echocardiographic changes in athletes include a 10-20% increase in LVWT and a 10-15% increase in both ventricular cavities dimensions compared to the similar age and weight population. Numerous studies were undertaken to ascertain the upper limits of physiological left ventricular hypertrophy (LVH) in athletes, leading to a consensus set at 10 mm for Caucasian women and 12 mm for Caucasian men, whereas in athletes of black African or Afro-Caribbean ethnicity, ventricular hypertrophy is greater and these limits were adjusted to 11 mm for women and 14 mm for men. The greatest changes in the Caucasian population are observed in male endurance athletes with a large body surface area, e.g. cross-country skiers, rowers, long-distance cyclists and runners, swimmers, soccer and tennis players [36-37]. These data were confirmed by CMRI studies [31]. However, only 2% of them achieve LVWT exceeding 12 mm [38] and among adolescent athletes, only 0.4% exceed 11 mm [39]. In extreme cases these values can raise diagnostic doubts whether to diagnose HCM or not, resulting in the so-called “gray zone”, comprising < 2% of athletes who present LVH higher than the acceptable norm for this population but below the value of 15 mm that meets the criteria for HCM [24]. In such cases, expanding the diagnosis with additional tests (e.g. ECG, CMRI, exercise testing) is sufficient to confirm or exclude the diagnosis of HCM [10]. The first step should be a reevaluation of the LVWT to exclude measurement error (for example, due to erroneous expansion of the dimension by trabeculae carneae, papillary muscles or the moderator band). This should be followed by the comparison of these findings with an ECG examination. This is because every athlete eventually diagnosed with mild HCM had an abnormal ECG in the past. Structural and functional parameters alone were not enough to distinguish them from a healthy population exhibiting hypertrophy due to physical activity. Also, the uniform pattern of LVH in the athlete’s heart differs from the asymmetric septal hypertrophy observed in 60% of HCM and apical hypertrophy in 10% of HCM. In contrast, LV cavity dilatation in athletes compensates for hypertrophy resulting in normal end-diastolic volume, which is typically reduced in HCM and compensated by hyperdynamic radial LV systolic function [3, 40].
Doppler evaluation of LV wall and diastolic mitral inflow patterns together with new methods allowing the precise evaluation of tissue distortion (e.g. measuring two-dimensional strain-derived velocity, strain, strain rate) can detect little disfunctions in long-axis relaxation which often occur a few years before the onset of obvious LVH in HCM. On the other hand, athletes often show above normal diastolic function, demonstrating high rates of early mitral inflow and early tissue relaxation by Doppler and high stroke volume even at elevated heart rates which, in combination with increased oxidative capacity and capillary conductance in skeletal muscle, translates into high VO2 max during exercise [8, 40]. A study comparing echocardiographic parameters of athletes and non-athletes with HCM reported higher diastolic filling values in the former group, most of which were within normal limits (93% of athletes had an E/E’ of < 12, 60% a septal E’ ≥ 0.09 m/s and 87% a lateral E’ ≥ 0.09 m). Hence, normal diastolic parameters and high VO2 max do not rule out the diagnosis of HCM in athletes [33].
In addition to reduced diastolic function and low VO2 max, features that may suggest a diagnosis of HCM include exercise-induced arrhythmias, reduced longitudinal systolic function and regional abnormalities of LV wall motion. The reduced longitudinal systolic function is assessed via global longitudinal strain (GLS) and is connected with an increased risk of heart failure (HF) even in the presence of a normal LV ejection fraction [41-42]. Although GLS at rest in athletes with HCM can be comparable to a control group of healthy athletes (who tend to have it slightly lowered), in general the average two-dimensional GLS in the group with HCM is reduced and its reduction in basal LV segments is a potential early indicator of HCM [43-45]. However, recent studies propose that changes in global and regional strain may not be useful in evaluating athletes, as they are a variant of the norm in this population [46].
In summary, an image eminently indicative of HCM in an athlete with LV hypertrophy (13-16 mm) would be a relatively small LV cavity (about 50 mm), hyperdynamic LV systolic function, dynamic LVOT stenosis during exercise, and in addition evidence of asymmetry of hypertrophy in regional/global strain (see Table 2) [8]. However, this clinical picture is quite rare given the variety of athletes’ hearts and we are usually dealing with equivocal findings that can be classified only after additional studies.
Table 2. The differences in the echocardiography in HCM and athlete’s heart
Cardiac magnetic resonance imaging
In the case of ECG changes and the absence of characteristic features or the presence of borderline abnormalities on echo, CMRI can more accurately reveal focal hypertrophy, especially of the posterior septum, apical and the free wall of the heart. Delayed enhancement after LGE, particularly in the region of the largest hypertrophies, can reveal typical features of cardiac fibrosis and influence the prognosis of HCM [31]. However, the lack of LGE does not rule out the diagnosis because it is not seen in as many as 60% of patients with HCM [47]. Additional changes may include elongation of the anterior mitral valve leaflet, the presence of an apical-basal LV myocardial bundle, abnormal position of the papillary muscles or myocardial crypts (see Table 3) [45]. These changes are suggestive of a diagnosis of HCM in suspected patients, particularly in family members carrying a pathogenic mutation without obvious changes in echo [48].
Table 3. Cardiac magnetic resonance imaging (CMRI) changes in HCM
Stress testing
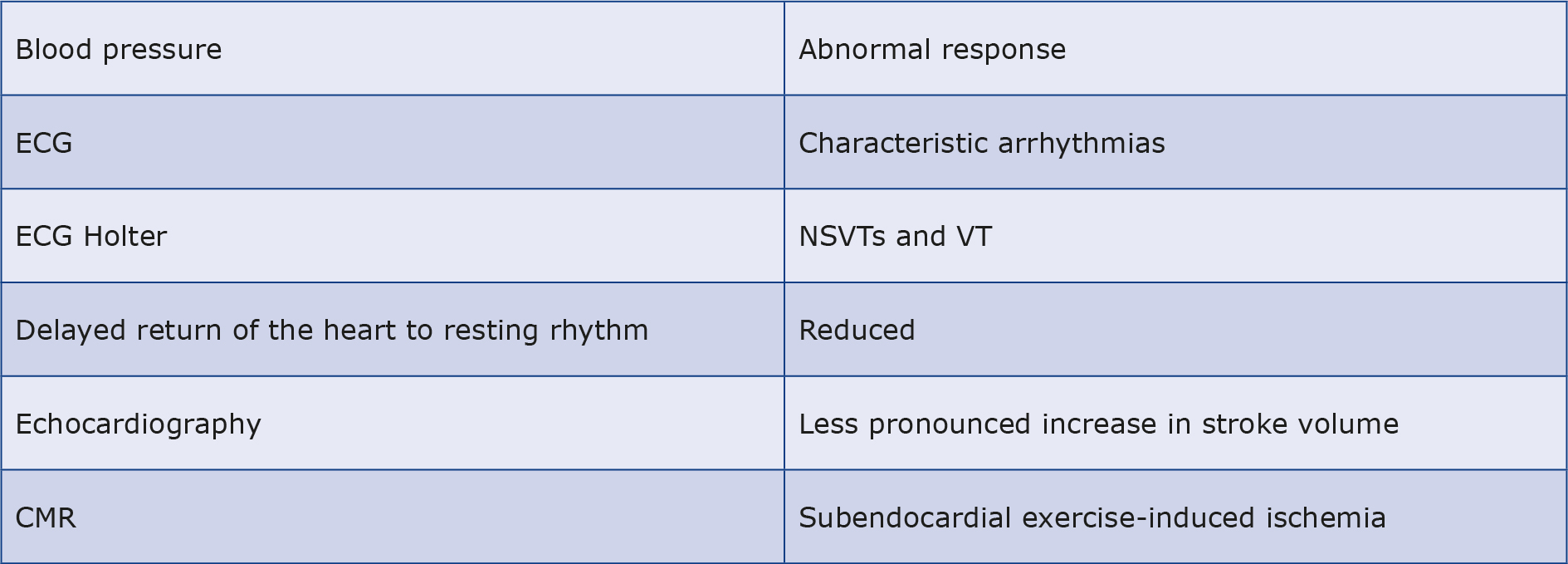
In the section on echo, it was shown how the dilatation of the heart cavities compensates for LVH in athletes and therefore high VO2 max is observed. Virtually all athletes with HCM are thus asymptomatic and their excellent exercise capacity does not rule out a diagnosis of HCM. Although a VO2 max > 50 ml/kg/min usually indicates an athlete’s heart [33, 49]. Nevertheless, increased physical activity may reveal characteristic arrhythmias, decreased GLS in an athlete with HCM, abnormal blood pressure response (flat curve or even a drop due to abnormal vascular tone combined with small vessel ischemia and exercise-induced LVOT stenosis [50]), delayed return of the heart to resting rhythm, increased LVOT gradient and less pronounced increase in stroke volume. The role of echo and CMRI stress testing is ancillary to concerns unresolved by other tests [51]. CMRI perfusion studies can also reveal areas of subendocardial exercise-induced ischemia, which never occurs in the athlete’s heart and is strongly suggestive of HCM (see Table 4) [52].
Table 4. Exercise-induced cardiac changes in HCM
Genetic testing
During initial testing, careful consideration should be given to a family history of sudden unexplained death, particularly in individuals < 50 years of age or documented cases of HCM within the family. If 1st degree relatives present with suspicious ECG changes this can also be a diagnostic clue [31]. Identification of a variant with some pathogenicity in a person with suspicious features on other tests can confirm the recognition of HCM and expedite similar diagnosis in other family members, but genetic testing should not be performed routinely [10], because in 40%-50% of patients with HCM the genetic basis is unknown and the various genetic mechanisms responsible for the pathophysiology of this disease remain largely enigmatic [53-54]. The diverse functions of these genes, e.g. encoding proteins that regulate calcium flow, building the cardiac sarcomere and Z-disc, influence the heterogeneity of the clinical picture of this disease. Moreover, it seems that the cardiac risk in gene-positive-phenotype-negative patients is similar to the population risk and current guidelines do not exclude such individuals from competitive sports after confirming the absence of abnormalities in echo and CMRI studies [5, 11]. In asymptomatic carriers of known pathogenic variants, LVH most often appears between 12 and 20 years of age and is not accompanied by any symptoms. Some of them will never present with full-blown HCM, and in some the disease may not appear until middle age [5, 55-56].
Discussion
Detraining
Since most athletes diagnosed with HCM are asymptomatic, no treatment is offered and they undergo follow-up, which provides a definitive opportunity to verify the diagnosis. Indeed, it has been proven that LVH associated strictly with prolonged, intense exercise regresses and after a 6-week period of training cessation the heart returns to a normal size range [57]. While there are also insights that reverse cardiac remodeling also occurs to some degree in athletes with HCM and is hard to distinguish from remodeling in healthy athletes; which in turn may be incomplete and persist despite long periods without activity [58-60]. It seems that further studies on larger groups will allow better decision-making about the complete suspension of training, which obviously has a significant impact on the life of an athlete.
Can sport cause cardiomyopathy?
There is one more issue that further complicates the picture of HCM in athletes. Namely, recently emerging studies that may support the development of secondary HCM due to the influence of intense exercise. A three-fold higher incidence of LGE has been demonstrated among experienced marathon runners, compared to the sedentary lifestyle population (12 vs. 4%), along with elevated markers of myocardial damage in this group, which is most likely related to a transient impairment of LV relaxation during increased cardiac action [61-63]. The significance of these parameters is not fully known, but a single small-group CMRI study conducted immediately after the marathon showed no obvious signs of myocarditis [64]. A study involving rats subjected to treadmill exercise for 16 weeks described LVH, diastolic dysfunction, and diffuse atrial and right ventricular fibrosis. In electrophysiological studies exercising rats also presented ventricular tachyarrhythmias more frequently than sedentary rats (42% vs. 6%) [65].
These results were not confirmed by a study on 114 Olympic athletes in endurance sports who intensively trained to 2-5 consecutive Olympic Games. No deterioration of cardiac function or arrhythmias was noted [66]. Moreover, it has been proven that people engaging in the most grueling sports have longer life expectancy than those with sedentary lifestyle, which may be related to the observed prevention of age-related decline in elasticity and compliance in physically active individuals, which predisposes to cardiovascular disease later in life [67-68].
It is also important to briefly mention the performance-enhancing substances (doping) that can disrupt the picture of myocardial damage in athletes. The most cardiotoxic of these substances are androgen anabolics, which cause LVH and LV dysfunction (including arrhythmias), which along with a worsening of lipid profile and elevated blood pressure can lead to myocardial infarction [69]. LVH and myocardial fibrosis can also be caused by human chorionic gonadotropin and human growth hormone [70].
Conclusions
Diagnosing HCM in athletes is complex due to the overlapping physiological changes from exercise. Risk assessment for SCD relies on comprehensive, multifaceted screening methods like family history, ECG, echo, CMRI, and stress testing. While strict guidelines advise limiting activity in diagnosed cases, it is crucial to individualize management, considering evolving research on exercise-induced cardiomyopathies. Furthermore, implementing universal athlete screening and considering genetic testing in suspicious cases could enhance early detection and preventive measures, thus mitigating the risk of SCD in this population. Genetic testing offers insights but its routine use is not recommended. Detraining may reverse some cases, showing the adaptability of heart muscle. Emerging studies suggest exercise-induced secondary cardiomyopathies, needing further investigation and evaluation in relation to the undeniable health benefits achieved through physical activity.
Conflict of interest
The authors declare that they have no conflicts of interest.
Funding
Not applicable.
--------------
Image – Patrick J. Lynch / echopedia.org(Creative Commons Attribution 2.5 License 2006)
References
| 1. |
Ciarambino T, Menna G, Sansone G, Giordano M. Cardiomyopathies: An Overview. Int J Mol Sci [Internet]. 2021;22(14):7722. Available from: https://www.mdpi.com/1422-0067/22/14/7722.
|
| 2. |
2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J [Internet]. 2014;35(39):2733–79. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehu284.
|
| 3. |
Cavalcante JL, Barboza JS, Lever HM. Diversity of Mitral Valve Abnormalities in Obstructive Hypertrophic Cardiomyopathy. Prog Cardiovasc Dis [Internet]. 2012;54(6):517–22. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0033062012000394.
|
| 4. |
Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic Cardiomyopathy. J Am Coll Cardiol [Internet]. 2014;64(1):83–99. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109714023390.
|
| 5. |
Maron BJ, Maron MS, Semsarian C. Genetics of Hypertrophic Cardiomyopathy After 20 Years. J Am Coll Cardiol [Internet]. 2012;60(8):705–15. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109712015860.
|
| 6. |
Kooreman Z, Giraldeau G, Finocchiaro G, Kobayashi Y, Wheeler M, Perez M, et al. Athletic Remodeling in Female College Athletes: The “Morganroth Hypothesis” Revisited. Clin J Sport Med [Internet]. 2019;29(3):224–31. Available from: https://journals.lww.com/00042752-201905000-00008.
|
| 7. |
Beaudry R, Haykowsky MJ, Baggish A, La Gerche A. A Modern Definition of the Athlete’s Heart—for Research and the Clinic. Cardiol Clin [Internet]. 2016;34(4):507–14. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0733865116300340.
|
| 8. |
Sharma S, Merghani A, Mont L. Exercise and the heart: the good, the bad, and the ugly. Eur Heart J [Internet]. 2015;36(23):1445–53. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehv090.
|
| 9. |
Pluim BM, Zwinderman AH, van der Laarse A, van der Wall EE. The Athlete’s Heart. Circulation [Internet]. 2000;101(3):336–44. Available from: https://www.ahajournals.org/doi/10.1161/01.CIR.101.3.336.
|
| 10. |
Brosnan MJ, Rakhit D. Differentiating Athlete’s Heart From Cardiomyopathies — The Left Side. Hear Lung Circ [Internet]. 2018;27(9):1052–62. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1443950618304682.
|
| 11. |
Maron BJ, Udelson JE, Bonow RO, Nishimura RA, Ackerman MJ, Estes NAM, et al. Eligibility and Disqualification Recommendations for Competitive Athletes With Cardiovascular Abnormalities: Task Force 3: Hypertrophic Cardiomyopathy, Arrhythmogenic Right Ventricular Cardiomyopathy and Other Cardiomyopathies, and Myocarditis. Circulation [Internet]. 2015;132(22). Available from: https://www.ahajournals.org/doi/10.1161/CIR.0000000000000239.
|
| 12. |
Maron BJ, Rowin EJ, Casey SA, Link MS, Lesser JR, Chan RHM, et al. Hypertrophic Cardiomyopathy in Adulthood Associated With Low Cardiovascular Mortality With Contemporary Management Strategies. J Am Coll Cardiol [Internet]. 2015;65(18):1915–28. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109715014175.
|
| 13. |
Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden Deaths in Young Competitive Athletes. Circulation [Internet]. 2009;119(8):1085–92. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.108.804617.
|
| 14. |
Maron BJ, Thompson PD, Ackerman MJ, Balady G, Berger S, Cohen D, et al. Recommendations and Considerations Related to Preparticipation Screening for Cardiovascular Abnormalities in Competitive Athletes: 2007 Update. Circulation [Internet]. 2007;115(12):1643–55. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.107.181423.
|
| 15. |
D’Ascenzi F, Valentini F, Pistoresi S, Frascaro F, Piu P, Cavigli L, et al. Causes of sudden cardiac death in young athletes and non-athletes: systematic review and meta-analysis. Trends Cardiovasc Med [Internet]. 2022;32(5):299–308. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1050173821000670.
|
| 16. |
Corrado D, Basso C, Schiavon M, Thiene G. Screening for Hypertrophic Cardiomyopathy in Young Athletes. N Engl J Med [Internet]. 1998;339(6):364–9. Available from: http://www.nejm.org/doi/abs/10.1056/NEJM199808063390602.
|
| 17. |
Harmon KG, Drezner JA, Wilson MG, Sharma S. Incidence of sudden cardiac death in athletes: a state-of-the-art review. Br J Sports Med [Internet]. 2014;48(15):1185–92. Available from: https://bjsm.bmj.com/lookup/doi/10.1136/bjsports-2014-093872.
|
| 18. |
Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol [Internet]. 2003;42(11):1959–63. Available from: https://linkinghub.elsevier.com/retrieve/pii/S073510970301194X.
|
| 19. |
Wren C. Cardiovascular pre‐participation screening of young competitive athletes for prevention of sudden death: proposal for a common European protocol. Eur Heart J [Internet]. 2005;26(17):1804–1804. Available from: http://academic.oup.com/eurheartj/article/26/17/1804/428594/Cardiovascular-preparticipation-screening-of-young.
|
| 20. |
Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in Sudden Cardiovascular Death in Young Competitive Athletes After Implementation of a Preparticipation Screening Program. JAMA [Internet]. 2006;296(13):1593. Available from: http://jama.jamanetwork.com/article.aspx?doi=10.1001/jama.296.13.1593.
|
| 21. |
Berdowski J, de Beus MF, Blom M, Bardai A, Bots ML, Doevendans PA, et al. Exercise-related out-of-hospital cardiac arrest in the general population: incidence and prognosis. Eur Heart J [Internet]. 2013;34(47):3616–23. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/eht401.
|
| 22. |
Kim JH, Malhotra R, Chiampas G, D’Hemecourt P, Troyanos C, Cianca J, et al. Cardiac Arrest during Long-Distance Running Races. N Engl J Med [Internet]. 2012;366(2):130–40. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMoa1106468.
|
| 23. |
Whyte G, Sheppard M, George K, Shave R, Wilson M, Prasad S, et al. Post-mortem evidence of idiopathic left ventricular hypertrophy and idiopathic interstitial myocardial fibrosis: is exercise the cause? Br J Sports Med [Internet]. 2008 Apr;42(4):304–5. Available from: https://bjsm.bmj.com/lookup/doi/10.1136/bjsm.2007.038158.
|
| 24. |
Basavarajaiah S, Boraita A, Whyte G, Wilson M, Carby L, Shah A, et al. Ethnic Differences in Left Ventricular Remodeling in Highly-Trained Athletes. J Am Coll Cardiol [Internet]. 2008;51(23):2256–62. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109708011157.
|
| 25. |
Corrado D, Pelliccia A, Heidbuchel H, Sharma S, Link M, Basso C, et al. Recommendations for interpretation of 12-lead electrocardiogram in the athlete. Eur Heart J [Internet]. 2010;31(2):243–59. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehp473.
|
| 26. |
Sheikh N, Papadakis M, Ghani S, Zaidi A, Gati S, Adami PE, et al. Comparison of Electrocardiographic Criteria for the Detection of Cardiac Abnormalities in Elite Black and White Athletes. Circulation [Internet]. 2014;129(16):1637–49. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.113.006179.
|
| 27. |
Zaidi A, Sharma S. Exercise and Heart Disease: From Athletes and Arrhythmias to Hypertrophic Cardiomyopathy and Congenital Heart Disease. Future Cardiol [Internet]. 2013;9(1):119–36. Available from: https://www.tandfonline.com/doi/full/10.2217/fca.12.81.
|
| 28. |
Pelliccia A, Maron BJ, Culasso F, Di Paolo FM, Spataro A, Biffi A, et al. Clinical Significance of Abnormal Electrocardiographic Patterns in Trained Athletes. Circulation [Internet]. 2000;102(3):278–84. Available from: https://www.ahajournals.org/doi/10.1161/01.CIR.102.3.278.
|
| 29. |
Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet [Internet]. 2017;389(10075):1253–67. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0140673616313216.
|
| 30. |
Sharma S, Whyte G, Elliott P, Padula M, Kaushal R, Mahon N, et al. Electrocardiographic changes in 1000 highly trained junior elite athletes. Br J Sports Med [Internet]. 1999;33(5):319–24. Available from: https://bjsm.bmj.com/lookup/doi/10.1136/bjsm.33.5.319.
|
| 31. |
Luijkx T, Cramer MJ, Prakken NHJ, Buckens CF, Mosterd A, Rienks R, et al. Sport category is an important determinant of cardiac adaptation: an MRI study. Br J Sports Med [Internet]. 2012;46(16):1119–24. Available from: https://bjsm.bmj.com/lookup/doi/10.1136/bjsports-2011-090520.
|
| 32. |
McLeod CJ, Ackerman MJ, Nishimura RA, Tajik AJ, Gersh BJ, Ommen SR. Outcome of Patients With Hypertrophic Cardiomyopathy and a Normal Electrocardiogram. J Am Coll Cardiol [Internet]. 2009;54(3):229–33. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109709013564.
|
| 33. |
Sheikh N, Papadakis M, Schnell F, Panoulas V, Malhotra A, Wilson M, et al. Clinical Profile of Athletes With Hypertrophic Cardiomyopathy. Circ Cardiovasc Imaging [Internet]. 2015;8(7). Available from: https://www.ahajournals.org/doi/10.1161/CIRCIMAGING.114.003454.
|
| 34. |
Papadakis M, Carre F, Kervio G, Rawlins J, Panoulas VF, Chandra N, et al. The prevalence, distribution, and clinical outcomes of electrocardiographic repolarization patterns in male athletes of African/Afro-Caribbean origin. Eur Heart J [Internet]. 2011;32(18):2304–13. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehr140.
|
| 35. |
Weissler-Snir A, Chan RH, Adler A, Care M, Chauhan V, Gollob MH, et al. Usefulness of 14-Day Holter for Detection of Nonsustained Ventricular Tachycardia in Patients With Hypertrophic Cardiomyopathy. Am J Cardiol [Internet]. 2016;118(8):1258–63. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002914916312528.
|
| 36. |
Pelliccia A. Physiologic Left Ventricular Cavity Dilatation in Elite Athletes. Ann Intern Med [Internet]. 1999;130(1):23. Available from: http://annals.org/article.aspx?doi=10.7326/0003-4819-130-1-199901050-00005.
|
| 37. |
Spirito P, Pelliccia A, Proschan MA, Granata M, Spataro A, Bellone P, et al. Morphology of the “athlete’s heart” assessed by echocardiography in 947 elite athletes representing 27 sports. Am J Cardiol [Internet]. 1994;74(8):802–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/0002914994904391.
|
| 38. |
Pelliccia A, Maron BJ, Spataro A, Proschan MA, Spirito P. The Upper Limit of Physiologic Cardiac Hypertrophy in Highly Trained Elite Athletes. N Engl J Med [Internet]. 1991;324(5):295–301. Available from: http://www.nejm.org/doi/abs/10.1056/NEJM199101313240504.
|
| 39. |
Sharma S, Maron BJ, Whyte G, Firoozi S, Elliott PM, McKenna WJ. Physiologic limits of left ventricular hypertrophy in elite junior athletes. J Am Coll Cardiol [Internet]. 2002;40(8):1431–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109702022702.
|
| 40. |
Teske AJ, Cox MG, De Boeck BW, Doevendans PA, Hauer RN, Cramer MJ. Echocardiographic Tissue Deformation Imaging Quantifies Abnormal Regional Right Ventricular Function in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J Am Soc Echocardiogr [Internet]. 2009;22(8):920–7. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0894731709004829.
|
| 41. |
Chandra N, Bastiaenen R, Papadakis M, Sharma S. Sudden Cardiac Death in Young Athletes. J Am Coll Cardiol [Internet]. 2013;61(10):1027–40. Available from: https://linkinghub.elsevier.com/retrieve/pii/S073510971205735X.
|
| 42. |
Hiemstra YL, Debonnaire P, van Zwet EW, Bootsma M, Schalij MJ, Bax JJ, et al. Development of and Progression of Overt Heart Failure in Nonobstructive Hypertrophic Cardiomyopathy. Am J Cardiol [Internet]. 2018;122(4):656–62. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002914918310397.
|
| 43. |
Schnell F, Matelot D, Daudin M, Kervio G, Mabo P, Carré F, et al. Mechanical Dispersion by Strain Echocardiography: A Novel Tool to Diagnose Hypertrophic Cardiomyopathy in Athletes. J Am Soc Echocardiogr [Internet]. 2017;30(3):251–61. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0894731716306769.
|
| 44. |
Kansal MM, Lester SJ, Surapaneni P, Sengupta PP, Appleton CP, Ommen SR, et al. Usefulness of Two-Dimensional and Speckle Tracking Echocardiography In “Gray Zone” Left Ventricular Hypertrophy to Differentiate Professional Football Player’s Heart from Hypertrophic Cardiomyopathy. Am J Cardiol [Internet]. 2011;108(9):1322–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002914911022454.
|
| 45. |
Peyrou J, Réant P, Reynaud A, Cornolle C, Dijos M, Rooryck-Thambo C, et al. Morphological and functional abnormalities pattern in hypertrophy-free HCM mutation carriers detected with echocardiography. Int J Cardiovasc Imaging [Internet]. 2016;32(9):1379–89. Available from: http://link.springer.com/10.1007/s10554-016-0929-6.
|
| 46. |
Swoboda PP, Erhayiem B, McDiarmid AK, Lancaster RE, Lyall GK, Dobson LE, et al. Relationship between cardiac deformation parameters measured by cardiovascular magnetic resonance and aerobic fitness in endurance athletes. J Cardiovasc Magn Reson [Internet]. 2016;18(1):48. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1097664723009894.
|
| 47. |
Quarta G, Aquaro GD, Pedrotti P, Pontone G, Dellegrottaglie S, Iacovoni A, et al. Cardiovascular magnetic resonance imaging in hypertrophic cardiomyopathy: the importance of clinical context. Eur Hear J - Cardiovasc Imaging [Internet]. 2018;19(6):601–10. Available from: https://academic.oup.com/ehjcimaging/article/19/6/601/4772575.
|
| 48. |
Maron MS, Maron BJ, Harrigan C, Buros J, Gibson CM, Olivotto I, et al. Hypertrophic Cardiomyopathy Phenotype Revisited After 50 Years With Cardiovascular Magnetic Resonance. J Am Coll Cardiol [Internet]. 2009;54(3):220–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109709015198.
|
| 49. |
Sharma S, Elliott PM, Whyte G, Mahon N, Virdee MS, Mist B, et al. Utility of metabolic exercise testing in distinguishing hypertrophic cardiomyopathy from physiologic left ventricular hypertrophy in athletes. J Am Coll Cardiol [Internet]. 2000;36(3):864–70. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109700008160.
|
| 50. |
Kawasaki T, Azuma A, Kuribayashi T, Akakabe Y, Yamano M, Miki S, et al. Vagal enhancement due to subendocardial ischemia as a cause of abnormal blood pressure response in hypertrophic cardiomyopathy. Int J Cardiol [Internet]. 2008;129(1):59–64. Available from: https://linkinghub.elsevier.com/retrieve/pii/S016752730701128X.
|
| 51. |
Tower-Rader A, Betancor J, Lever HM, Desai MY. A Comprehensive Review of Stress Testing in Hypertrophic Cardiomyopathy: Assessment of Functional Capacity, Identification of Prognostic Indicators, and Detection of Coronary Artery Disease. J Am Soc Echocardiogr [Internet]. 2017;30(9):829–44. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0894731717303954.
|
| 52. |
Petersen SE, Jerosch-Herold M, Hudsmith LE, Robson MD, Francis JM, Doll HA, et al. Evidence for Microvascular Dysfunction in Hypertrophic Cardiomyopathy. Circulation [Internet]. 2007;115(18):2418–25. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.106.657023.
|
| 53. |
Marian AJ, Braunwald E. Hypertrophic Cardiomyopathy. Circ Res [Internet]. 2017;121(7):749–70. Available from: https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.117.311059.
|
| 54. |
Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, et al. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. J Card Fail [Internet]. 2018;24(5):281–302. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1071916418301015.
|
| 55. |
Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary. Circulation [Internet]. 2011;124(24):2761–96. Available from: https://www.ahajournals.org/doi/10.1161/CIR.0b013e318223e230.
|
| 56. |
Maron BJ, Niimura H, Casey SA, Soper MK, Wright GB, Seidman J., et al. Development of left ventricular hypertrophy in adults with hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. J Am Coll Cardiol [Internet]. 2001;38(2):315–21. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109701013869.
|
| 57. |
Spence AL, Naylor LH, Carter HH, Buck CL, Dembo L, Murray CP, et al. A prospective randomised longitudinal MRI study of left ventricular adaptation to endurance and resistance exercise training in humans. J Physiol [Internet]. 2011;589(22):5443–52. Available from: https://physoc.onlinelibrary.wiley.com/doi/10.1113/jphysiol.2011.217125.
|
| 58. |
de Gregorio C, Speranza G, Magliarditi A, Pugliatti P, Andò G, Coglitore S. Detraining-related changes in left ventricular wall thickness and longitudinal strain in a young athlete likely to have hypertrophic cardiomyopathy. J Sports Sci Med [Internet]. 2012;11(3):557–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24149368.
|
| 59. |
Ghani S, Sharma S. Electrocardiographic changes in an athlete before and after detraining. Case Reports [Internet]. 2012;2012(may07 1):bcr0120125520–bcr0120125520. Available from: https://casereports.bmj.com/lookup/doi/10.1136/bcr.01.2012.5520.
|
| 60. |
Pelliccia A, Maron BJ, De Luca R, Di Paolo FM, Spataro A, Culasso F. Remodeling of Left Ventricular Hypertrophy in Elite Athletes After Long-Term Deconditioning. Circulation [Internet]. 2002;105(8):944–9. Available from: https://www.ahajournals.org/doi/10.1161/hc0802.104534.
|
| 61. |
Oxborough D, Whyte G, Wilson M, O’Hanlon R, Birch K, Shave R, et al. A Depression in Left Ventricular Diastolic Filling following Prolonged Strenuous Exercise is Associated with Changes in Left Atrial Mechanics. J Am Soc Echocardiogr [Internet]. 2010;23(9):968–76. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0894731710004633.
|
| 62. |
Shave R, Baggish A, George K, Wood M, Scharhag J, Whyte G, et al. Exercise-Induced Cardiac Troponin Elevation. J Am Coll Cardiol [Internet]. 2010;56(3):169–76. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109710017055.
|
| 63. |
Breuckmann F, Möhlenkamp S, Nassenstein K, Lehmann N, Ladd S, Schmermund A, et al. Myocardial Late Gadolinium Enhancement: Prevalence, Pattern, and Prognostic Relevance in Marathon Runners. Radiology [Internet]. 2009;251(1):50–7. Available from: http://pubs.rsna.org/doi/10.1148/radiol.2511081118.
|
| 64. |
O’Hanlon R, Wilson M, Wage R, Smith G, Alpendurada FD, Wong J, et al. Troponin release following endurance exercise: is inflammation the cause? a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson [Internet]. 2010;12(1):38. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1097664723013182.
|
| 65. |
Benito B, Gay-Jordi G, Serrano-Mollar A, Guasch E, Shi Y, Tardif J-C, et al. Cardiac Arrhythmogenic Remodeling in a Rat Model of Long-Term Intensive Exercise Training. Circulation [Internet]. 2011;123(1):13–22. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.110.938282.
|
| 66. |
Pelliccia A, Kinoshita N, Pisicchio C, Quattrini F, DiPaolo FM, Ciardo R, et al. Long-Term Clinical Consequences of Intense, Uninterrupted Endurance Training in Olympic Athletes. J Am Coll Cardiol [Internet]. 2010;55(15):1619–25. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109710004493.
|
| 67. |
Marijon E, Tafflet M, Antero-Jacquemin J, El Helou N, Berthelot G, Celermajer DS, et al. Mortality of French participants in the Tour de France (1947-2012). Eur Heart J [Internet]. 2013;34(40):3145–50. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/eht347.
|
| 68. |
Bhella PS, Hastings JL, Fujimoto N, Shibata S, Carrick-Ranson G, Palmer MD, et al. Impact of Lifelong Exercise “Dose” on Left Ventricular Compliance and Distensibility. J Am Coll Cardiol [Internet]. 2014;64(12):1257–66. Available from: https://linkinghub.elsevier.com/retrieve/pii/S073510971404546X.
|
| 69. |
Luijkx T, Velthuis BK, Backx FJG, Buckens CFM, Prakken NHJ, Rienks R, et al. Anabolic androgenic steroid use is associated with ventricular dysfunction on cardiac MRI in strength trained athletes. Int J Cardiol [Internet]. 2013;167(3):664–8. Available from: https://linkinghub.elsevier.com/retrieve/pii/S016752731200277X.
|
| 70. |
Deligiannis A, Björnstad H, Carre F, Heidbüchel H, Kouidi E, Panhuyzen-Goedkoop NM, et al. ESC Study Group of Sports Cardiology Position Paper on adverse cardiovascular effects of doping in athletes. Eur J Cardiovasc Prev Rehabil [Internet]. 2006;13(5):687–94. Available from: https://academic.oup.com/eurjpc/article/13/5/687-694/5933267.
|

{kind=link}