
Advances in understanding, diagnosis and targeting ATTR cardiomyopathy: a review


Abstract
Transthyretin amyloidosis (ATTR) manifests as wild-type (ATTRwt) and hereditary/mutant (ATTRv) forms and can lead to heart failure due to cardiac amyloidopathy. Diagnosing ATTR, particularly in asymptomatic carriers of pathogenic variants, remains challenging despite the advances. Complex and multi-aspect management involves a limited range of well-examined conventional therapies to address the heart failure and frequently coexisting arrhythmias and valvular issues. Disease-modifying treatment, RNA-based treatments, CRISPR-Cas9 gene editing and monoclonal antibodies targeting amyloid deposits are recent and promising innovations. This review explores the diagnostic intricacies, therapeutic dilemmas and emerging solutions in ATTR cardiomyopathy. The significance of early detection and precise, targeted approaches to enhance patient outcomes is underscored.
Citation
Kulak M, Lang M, Waśniewski M, Lange N. Advances in understanding, diagnosis and targeting ATTR cardiomyopathy: a review. Eur J Transl Clin Med. 2024;7(1):63-78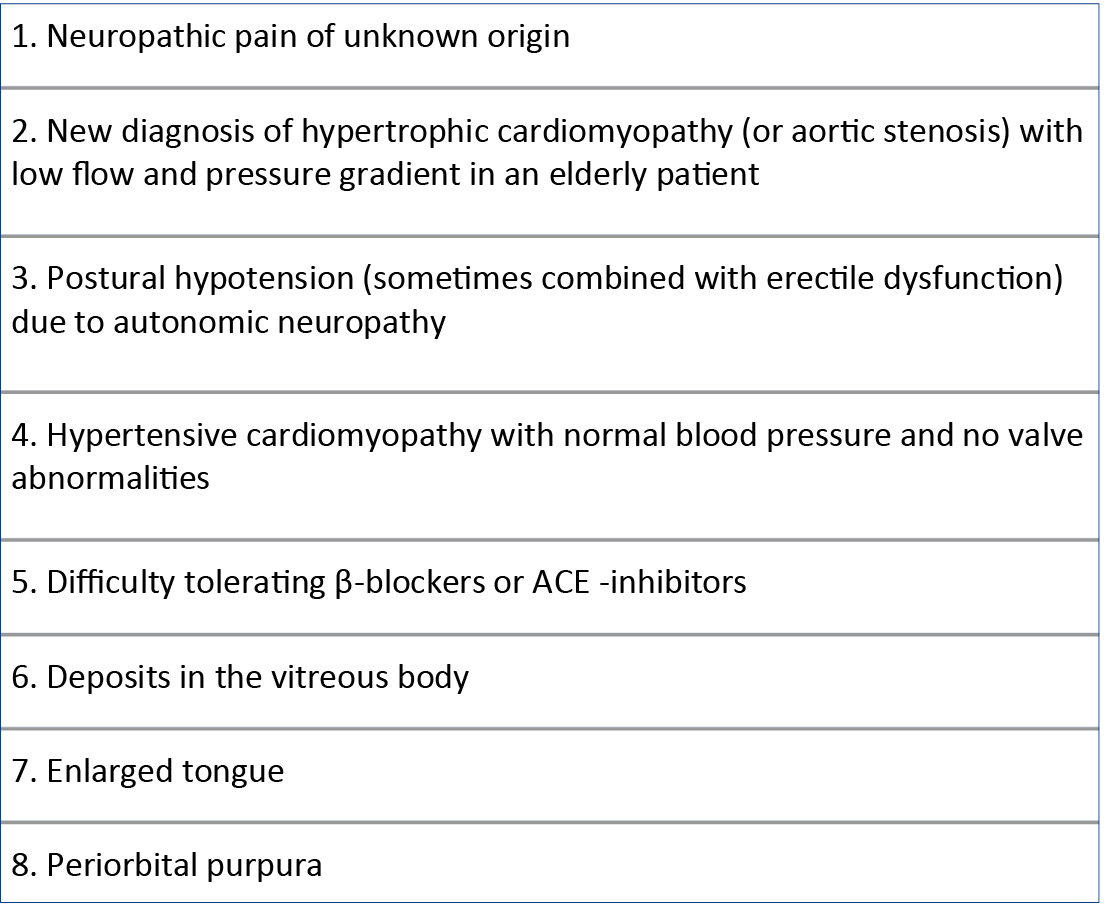




Introduction
Amyloidosis is a diverse group of diseases whose pathogenesis involves the deposition of insoluble protein deposits of various origins that impair the function of internal organs, including the heart. Vast majority (95%) of cardiac amyloidosis (CA) cases are light chain amyloidosis (AL) and transthyretin amyloidosis (ATTR) [1]. These are rare diseases, but their estimated prevalence in the European population is 3/1000 [2] and in recent years the incidence of ATTR-CA has increased several-fold [3] and might be underestimated [4-5]. Transthyretin (TTR) is a protein produced in the liver, involved in the transport of thyroid hormones and vitamin A in the blood, its defective spatial conformation causes the formation of deposits in ATTR.
ATTR is divided into the wild type (ATTRwt) associated with aging and the mutant or hereditary type (ATTRv), which is characterized by a single amino acid substitution in the chain of 127 amino acids that make up the protein. It can occur familially, although up to 50% of patients with this type have no confirmed cases among relatives [6-7].
ATTRv with polyneuropathy is estimated to affect 5000- -10000 people worldwide [8]. It can be divided into late-onset (after age 50) and early-onset ATTRv, the former of which is usually sporadic and has a more aggressive course with predominant peripheral neuropathy [9]. More than 130 pathogenic variants of TTR are known [6]. Some variants (e.g. p.Leu131Met) manifest complete penetrance in a specific age range, but most have incomplete penetrance [3, 10] and the chance of revealing clinical features increases with age [11]. The most common variant is p.V142I – named after the amino acid substitution site. Carriers of this mutation are 3-4% of the global population of African ancestry and in the United States it may account for about 23% of ATTR-CA cases [12]. Therefore, it is suspected that it may be a founder mutation or codon 142 could be a mutational hotspot [3, 13]. In European population-based studies, the frequency of the mutant type among patients with ATTR-CA was just a few percent [3, 13]. Most patients with ATTRv were of African and Caribbean descent and usually carried the p.V142I mutation [13-14].
The hereditary form of ATTR is distinguished by the onset of symptoms at a younger age [3] and usually a co-occurrence of cardiac and neurological symptoms [15]. At the same time, the occurrence of ATTRv is associated with increased mortality and a more severe course of the disease [16]. Hence, genetic screening tests for ATTRv have been advocated for all patients with a diagnosis of ATTR and genetic testing was included in the diagnostic protocol for hypertrophic cardiomyopathy [17-18], whose clinical picture may resemble ATTR-CA.
Wild-type ATTR is more common and associated with aging, usually affecting white males around the age of 80, hence it is sometimes called “senile amyloidosis.” Cardiac manifestation (restrictive cardiomyopathy with diastolic heart failure) in this type of AL is often overlooked and underestimated due to the overlap of concomitant diseases in this age group, e.g. hypertension, valvular defects, heart failure (HF) due to ischemic disease, hypertrophic cardiomyopathy. ATTR-CA was detected in about 13% of patients hospitalized with HFpEF (heart failure with preserved ejection fraction) and about 16% with severe aortic stenosis undergoing transcatheter aortic valve implantation (TAVI) [6, 19]. Diagnostic vigilance should be increased when other systemic manifestations of amyloidosis occur, often preceding cardiac involvement by several years [20], e.g. carpal tunnel syndrome (almost always bilateral), spinal canal stenosis or rupture of the biceps tendon [5-6].
The aim of our study was to explore the diagnostic intricacies, therapeutic dilemmas, and emerging solutions in ATTR cardiomyopathy, underscoring the significance of early detection and precise, targeted approaches to enhance patient outcomes.
Materials and methods
A comprehensive literature search in PubMed and Google Scholar was conducted to identify relevant studies pertaining to ATTR cardiomyopathy. The search was performed using electronic databases including. The following search terms were used: “ATTR cardiomyopathy,” “transthyretin,” “diagnosis,” “amyloidosis,” “treatment,” “genetic mutations,” and “clinical trials” and their equivalents in Polish. The search was limited to articles published in English and Polish. Studies were included if they focused on any aspect of ATTR cardiomyopathy, including its pathophysiology, clinical manifestations, diagnosis, treatment modalities, genetic mutations and recent advances. Both human and animal studies were considered. After excluding 57 abstracts that were duplicated or not directly related to ATTR cardiomyopathy or amyloidosis, a total of 93 reviews, case reports, original research articles and clinical trials were included in this review.
Results and discussion
Pathogenesis of myocardial damage
Deposition of TTR occurs most often interstitially (around the myocytes) and leads to restrictive cardiomyopathy (due to increased stiffness and thickness of both chambers of the heart) [4]. If the deposits are more intramural, occupying the interventricular or interatrial septum, they may also cause features of hypertrophic cardiomyopathy, ultimately leading to HFpEF [15]. Due to low wall compliance, relatively higherincoming blood pressure is needed to fill the ventricles, leading to left ventricular (LV) diastolic failure [21]. During the progression of the disease, stroke volume, compliance and minute volume gradually decrease [6], resulting in increased mortality [22]. Furthermore, there is laboratory evidence that TTR oligomers induce specific cellular abnormalities to the cultured cardiomyocytes leading to reduction in their survival time and may lead to alterations within the electrical function of cardiomyocytes resulting in arrhythmias [23].
Cardiac involvement at diagnosis is usually asymptomatic. The first symptoms of the disease are nonspecific, therefore can contribute to misdiagnosis e.g. exertional dyspnea and atrial fibrillation (with possible embolic complications) lower limb edema and ascites [4-5].
Atrial fibrillation
The most common persistent arrhythmia in ATTR-CA is atrial fibrillation (AF) [6]. It is more common in ATTRwt (incidence 27-71%) than in ATTRv (5-28%) [24] and is characterized by a higher propensity for thrombosis than in the general population. Up to about 30% of the patients had a cardiac thrombus [25] of which up to 87% were taking anticoagulants, thus marginalizing the role of the CHA2DS2-VASc score in this group of patients [26] and suggesting the use of systemic anticoagulation in all cases of AF in ATTR-CA. Also, patients with preserved sinus rhythm in ATTR-CA have a higher risk of embolization due to impaired left atrial function [4, 27].
No differences in survival have been demonstrated between patients with pharmacologically restored sinus rhythm (amiodarone) and those treated according to the heart rate (HR) control strategy [28]. Sinus rhythm restoration and ablation are more effective when used in the initial phase of the CA [29-30]. Despite the efficacy of cardioversion being comparable to other groups, the rate of AF recurrence after one year in this group of patients is high [31]. Also, recurrences after ablation are common (58% recurrence rate after 39 months) [29].
Regarding HR rate control strategy, cautious use of beta- -blockers in persistent AF or non-dihydropyridine calcium channel blockers is recommended at the lowest possible dose and with frequent monitoring of the patient’s condition due to the risk of hypotension and potential enhancement of amyloid accumulation [24]. A possible alternative in patients at high risk of hypotension is digoxin [32]. It should be remembered that the use of digoxin and calcium blockers is contraindicated if the LV outflow tract is narrowing in the course of hypertrophic cardiomyopathy.
Bradyarrhythmias
Amyloid deposition can cause conduction system dysfunction, ranging from atrio-ventricular (A-V) block I° (20%) to III°. Since left bundle branch block and complete heart block are more common in ATTR-CA than in AL-CA [4], pacemakers are routinely implanted in ATTRwt, but it has proven reasonable to implant them also in patients with familial amyloidosis who have a high risk of A-V block: bundle-branch His-ventricular (HV) interval ≥ 70 ms, HV interval > 55 ms (if associated with bundle-branch block), Wenckebach point ≤ 100 beats/ min [33]. In patients with particularly high pacing rates, who are at risk of pacing-induced cardiomyopathy [34] (which would worsen HF), cardiac resynchronisation therapy implantation can reduce HF symptoms while increasing left ventricular ejection fraction (LVEF) and survival [35].
Ventricular tachycardias
Cardiac death in the setting of ATTR-CA is rare [6]. Implantable cardioverter-defibrillator (ICD) implantation should be considered in patients with syncope and complex nonfixed ventricular arrhythmias [36], although many reports do not demonstrate increased survival compared with CA patients without ICDs [24]. The European Society of Cardiology (ESC) recommendations state that there is insufficient evidence for the efficacy of ICD use in the primary prevention of sudden cardiac death in CA [37]. The decision about ICD implantation should be made individually for every patient, particularly in patients with syncope of unknown origin and suspected proarrhythmogenic state [38].
Valvular diseases
Aortic stenosis often co-occurs with ATTR-CA, particularly in older men, who are at risk for both diseases. The best treatment option is TAVI, which has a lower procedural risk than traditional surgical valve replacement (AVR) [19, 39]. Unfortunately, patients with CA after TAVI have a higher risk fof hospitalization for HF [40] and an almost 2 times higher risk of death within 1.7 years compared to patients without additional burden [19].
Mitral and tricuspid valves are typically involved in ATTR-CA, often thickened and remodeled together with the interatrial septum [41], however the valve disease is most often hemodynamically insignificant. In a study of tissues obtained during mitral valve surgery, < 1% showed coexistence of ATTR-CA (more often ATTRwt type) [42]. In studies of small groups, percutaneous surgery was not associated with an increased risk of complications in these patients [43]. Notwithstanding, the importance of corrective interventions for tricuspid regurgitation is controversial, as it appears to be secondary to RV remodeling rather than resulting from primary valve dysfunction [44].
Transplantation as a therapeutic option
The majority of plasma TTR is synthesized in the liver, making orthotopic liver transplantation (OLT) a treatment option for TTR amyloidosis to halt the production of variant TTR in the blood. This procedure was reported beneficial for patients with the Val30Met mutation and neuropathy, particularly when qualified for the procedure in well-nourished state, early-stage and early-onset of the disease. However, clinical reports have indicated that cardiomyopathy and neuropathy can still progress in patients with TTR mutations other than Val30Met, as well as in some Val30Met patients following OLT. This information suggests that even after OLT, wild-type TTR produced by the new liver can still accumulate in the heart as amyloid. In cases where a patient undergoes combined heart and liver transplantation, amyloid deposition in the heart graft typically does not occur. However, there have been observations of progression of amyloid deposition in other organ systems following this procedure, making the genetic screening important prognostic tool for identification of higher risk patients with TTRwt mutation [33, 45-46].
Disease-modifying treatment
The latest treatment strategies for ATTR-CA are based on two mechanisms: spatial stabilization of TTR and silencing of the genes responsible for its production [47]. The group of stabilizing drugs includes Diflunisal, Tafamidis and Acoramidis. The former is a non-steroidal anti-inflammatory drug that binds to the tetraiodothyronine binding site on the TTR tetramer, thereby limiting its abnormal folding into amyloid by slowing its dissociation time. When used at a low dose (250mg twice daily), it is usually well tolerated and results in slowing the progression of HF in ATTR-CA [48]. Tafamidis is a benzoxazole derivative, used in oral form, that reduces allcause mortality by 13.4% and the risk of hospitalization for cardiovascular reasons in patients in the New York Heart Association (NYHA) Classification class I or II by 32% at 30-month follow-up. The greatest mortality reduction benefit was demonstrated at 18 months after treatment implementation. Unfortunately, patients in NYHA class III showed an increased risk of hospitalization compared to placebo, while other side effects were not observed [49-50]. In preclinical trials there is also Acoramidis, which resembles in structure the p.T139M variant of TTR, which in patients who are heterozygotes for the p.V50M showed a protective effect and prevented familial amyloid polyneuropathy [51]. It seems that it could be more effective than the above-mentioned drugs, because it binds TTR in a more selective manner, provides stronger stabilization of both the p.V142I TTR variant and TTRwt [52].
Two genetic methods of silencing TTR production are available: antisense oligonucleotides (ASOs) and short interfering RNAs (siRNAs). The ASOs bind to proteins in the serum, on the cell surface and in its interior, where they start a chain of messenger ribonucleic acid (mRNA) degradation by incorporation with the target mRNA in the nucleus and activation of endonucleases [53]. Examples of ASOs include Inotersen and AKCEA-TTR-LRx. The former, which is a first-generation drug administered by weekly subcutaneous injections, stabilizes neuropathy and improves quality of life in ATTRv patients with polyneuropathy, regardless of myocardial involvement. As for the effect on the heart, during the 2-year follow-up an improvement in exercise tolerance in the 6-minute walk test by 20.2 meters and a decrease in mean LV mass in cardiac magnetic resonance (CMR) imaging by 8.4% were observed, and these positive results persisted after another year of follow-up [54]. However, the side effects of this drug require expanded studies, as it has been shown that it can induce glomerulonephritis and severe thrombocytopenia (PLT < 25,000/mL) in up to 6% of subjects [55], while the NEURO-TTR study (NCT01737398) did not confirm these reports and tolerability in the study group was good. AKCEA-TTR-LRx, on the other hand, is a second-generation drug, which in a phase 1 study (NCT03728634) proved more effective in lowering serum TTR levels by 85.7% (51-fold higher efficacy with 27-fold lower drug dose), what’s more, it can be administered by monthly subcutaneous injections and so far has not shown serious side effects [56].
The siRNAs are double-stranded oligonucleotides with a sense strand acting as a drug carrier and an antisense strand constituting the active molecule. The antisense strand binds to the target sequence present in all types of ATTR and forms a complex that silences the gene sequence in the process of RNA interference with subsequent degradation of TTR mRNA in the liver and reduction of TTR concentration in plasma [33,53]. The first generation of siRNAs is Patisiran, which reduces TTR levels in both wild-type and mutant ATTR [57]. Clinical benefits achieved after 18 months of use include reduced NT-proBNP, global longitudinal strain (GLS) and mean LV wall thickness, in addition to a 46% reduction in hospitalization and all-cause mortality compared with the placebo group. Unfortunately Patisiran has not been approved for the treatment ATTR-CA, because it has not yet been shown to influence cardiovascular outcomes in the other studies [58]. The second-generation drug in this group is Vutrisiran, which in the long-term (for 90 days) reduced TTR levels by 83% 6 weeks after use in phase 3 clinical trials (NCT04153149 and NCT03759379) which will be completed in December 2026 [59].
In 2018, the United States Food and Drug Administration approved both Patisiran and Inotersen for the treatment of genetically mediated ATTR polyneuropathy. Initial data from the studies mentioned above, were obtained from patients with ATTR polyneuropathy who also had symptoms of cardiomyopathy, suggest that we may expect equally beneficial treatment effects in patients with ATTR-related heart disease. Consequently, studies of both drugs are ongoing in phase 3 clinical trials in the population of patients with ATTR-related heart disease. Tafamidis is the first and so far the only drug with documented efficacy in a randomized study for treating ATTR-related heart disease. In the 2021 ESC guidelines regarding the diagnosis and treatment of acute and chronic HF, Tafamidis is designated as a Class I medication for ATTR-CA. Inotersen and Patisiran are recommended for consideration in cases of genetically mediated ATTR polyneuropathy [60].
Diagnostic problems
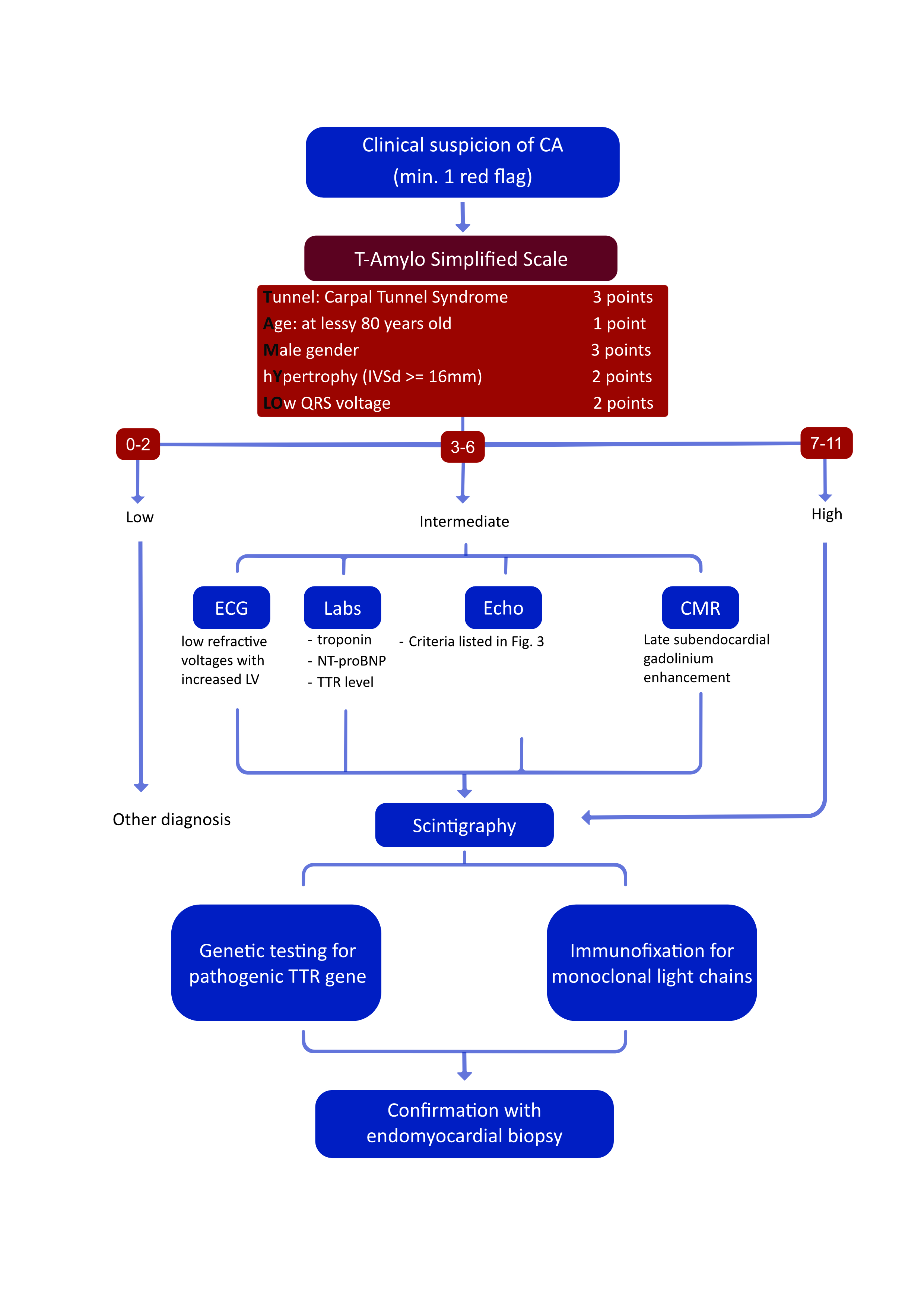
Since ATTR-CA manifests late, usually already in the advanced stage of HF, when treatment is no longer effective, much emphasis is placed on diagnostic possibilities in asymptomatic patients from at-risk groups (e.g. carriers of pathogenic variants with incomplete penetrance [6]) or in the early stage of the disease. As described above, some extracardiac manifestations of ATTR can be helpful in guiding towards the appropriate diagnostic pathway for CA, but there are other symptoms that can serve as “red flags” for the diagnosis of ATTR-CA: intolerance to beta-blockers or angiotensin-converting-enzyme inhibitors (ACEIs), low or normal blood pressure in hypertensive patients, a new diagnosis of hypertrophic cardiomyopathy or aortic stenosis with low flow and gradient in an elderly patient [4] (Figure 1). Recent studies have proven both the positive and the negative predictive value of some of the parameters mentioned and proposed the T-AMYLO scale simplifying the diagnostic process of ATTR-CA by identifying high-risk patients requiring further diagnostics and allowing ATTR-CA to be excluded in up to 30% of suspected patients with LV hypertrophy without the need for performing additional imaging tests (Figure 2). This seems to be a step towards non-invasive screening of patients at risk [40]. Known markers of HF, e.g. troponins (cTn) or N-terminal prohormone of brain natriuretic peptide, can be helpful for staging assessment and have been included in the new ATTR staging guideline [16], but do not identify asymptomatic carriers of TTRv in whom they tend to remain normal [61].
Figure 1. “Red flags” suggesting the diagnosis of ATTR-CA
Figure 2. Simplified ATTR-CA diagnostic algorithm
Adipose tissue biopsy can be used as an adjunct method in the diagnosis of ATTR-CA, but this method is invasive, has limitations related to the diverse localization of amyloid and does not definitively determine the etiology of CA [62].
The electrocardiogram (ECG) has negligible diagnostic [63] and prognostic value [6] in ATTR-CA, although low refractive voltages with increased LV wall thickness may suggest amyloidosis, which is more characteristic of AL-CA [64].
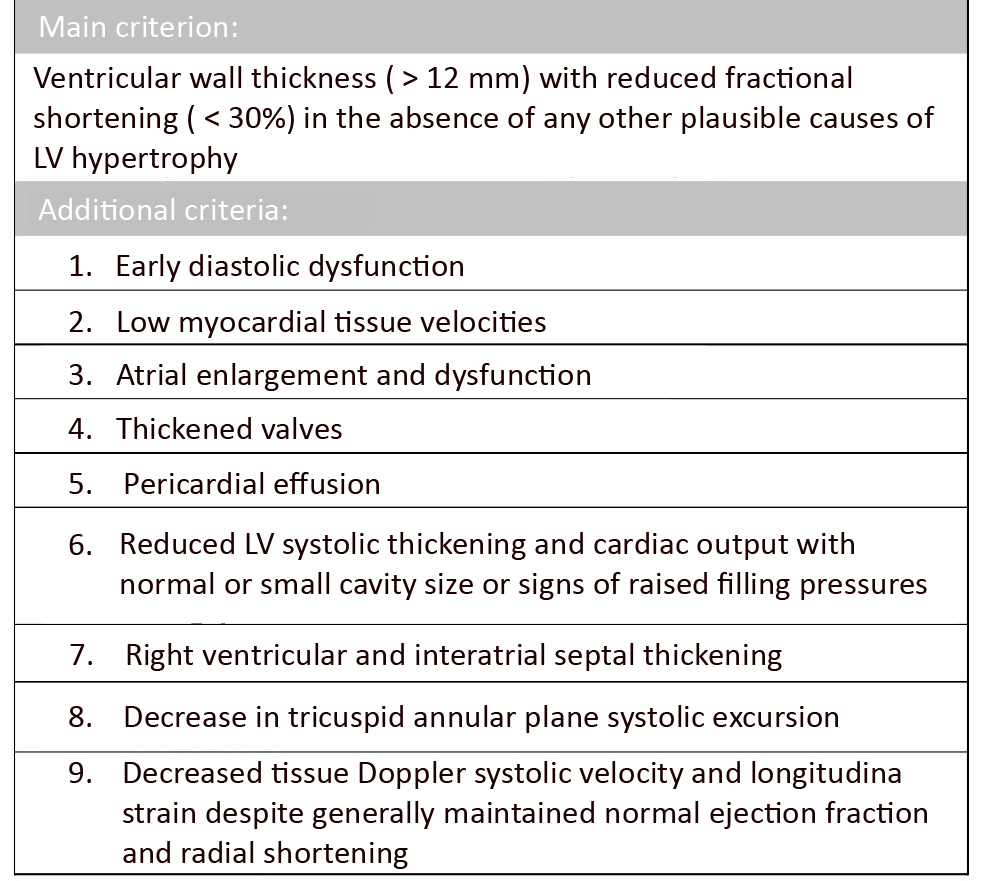
Echocardiography (echo) with Doppler assessment of myocardial strain is currently the leading method for assessing the progression and diagnosis of ATTR-CA even though it is not sufficient to alone diagnose CA [11, 65]. Some authors suggest over-reliance on planar imaging (echo, CMR), particularly in the context of asymptomatic patients in whom cardiac lesions may not be present or detectable [65]. Although no formal system for evaluating echocardiographic parameters in ATTR-CA has been established and lesions do not clearly distinguish ATTR from AL [6], several useful negative prognostic parameters have been identified. Of particular diagnostic importance is typical ventricular wall thickness (> 12mm) with reduced fractional shortening (< 30%) in the absence of any other plausible causes of LV hypertrophy [66] leading to early diastolic dysfunction with low myocardial tissue velocities. Other suggestive features are biatrial enlargement and dysfunction, thickened valves, pericardial effusion, reduced LV systolic thickening and reduced cardiac output with normal or small cavity size or signs of increased filling pressures (Figure 3).
Figure 3. Echocardiographic parameters of ATTR amyloidosis
The right ventricle is commonly influenced by both the direct presence of amyloid in its tissue and the increased pressure from pulmonary hypertension, leading to right ventricular and interatrial septal thickening, a decrease in all 3 parameters: TAPSE, tissue Doppler systolic velocity and LS, of which decreased LS is most typical feature of CA in a tissue Doppler imaging. Despite these deviations ejection fraction is generally maintained and there are no variations in radial shortening [65]. In both AL-CA and ATTR-CA, despite preserved LVEF, there is an early reduction of LS in the basal and middle segments of the heart cavity with typical sparing of the apex, which is not present in other causes of increased LV wall thickness [4]. Based on this feature, an equation was developed to distinguish ATTR-CA:
mean LS of the apex
________________________________________________
(mean LS of the base + LS of the center of the left ventricle
a result of 1.0 is the cutoff value for ATTR-CA [67]. To increase specificity, a multi-parameter diagnosic criteria has been proposed that includes, in addition to GLS with typical apical sparing and relative wall thickness, TAPSE and ratio between early mitral inflow velocity and mitral annular early diastolic velocity (E/E’) assessment [4].
CMR is a useful adjunct method to echo, in which the most diagnostically relevant is late subendocardial gadolinium enhancement (LGE) indicative of amyloid deposits that increase extracellular volume [65, 68]. CMR with LGE has a 93% sensitivity in detecting CA, but does not differentiate AL from ATTR-CA. Myocardial T1 mapping, a pixel-based reconstruction of measured longitudinal relaxation times, supplements the use of LGE in diagnosing CA via CMR. Besides its diagnostic usefulness, it can be utilized for monitoring and tracking myocardial amyloid infiltration and thus disease severity. Unlike LGE, native myocardial T1 (a measure of T1 time before the administration of contrast) offers an objective quantitative measurement rather than a subjective qualitative one. Native T1 demonstrates similar diagnostic performance in both AL and ATTR CA, often showing elevation in the early stages of CA before the onset of biventricular thickening or detectable LGE [69].
In AL-CA, testing for free light chains in serum is a highly sensitive biomarker, however at this time there are no tests based on TTR oligomer blood levels. However, high free light chain concentration might not be specific for AL-CA, particularly in patients over 65 years of age. Monoclonal gammopathy of undetermined significance (MGUS) affects up to 5% of people in this age group, potentially leading to misdiagnosis with ATTR-CA. Also, in chronic kidney disease, increased free light chains from impaired renal filtration might mask ATTR-CA. Coming back to TTR, its normal range in the serum is 18-45mg/dl and depends on age, sex, ethnicity and nutritional status. It can also vary due to infectious processes and inflammatory states (it correlated with c-reactive protein, sedimentation rate, albumin levels). More importantly, regardless of these factors, lower levels of TTR have been shown to be associated with increased mortality and worsening cardiac function in ATTRwt suggesting that it can be useful as disease progression indicator. In the same study 1- and 2-year follow-up was conducted in which the comparison was made between patients treated and not treated with diflunisal. Unchanged TTR in the not treated group corresponded with increased cTn-I (cardiac troponin I) and decreased LVEF [4, 70-71].
Differentiation with AL-CA
In the entire initial diagnostic process of CA, it is crucial to consider its most common subtype (70-80%), which is AL. This is particularly important due to the difficulty (or rather the inability) to differentiate it from ATTR-CA in basic imaging studies such as echo or CMR, as mentioned earlier. The misdiagnosis rates between hereditary TTR and AL amyloidosis can reach 7%, with the catastrophic possibility of performing a bone marrow transplant instead of an OLT or vice versa. Furthermore, the course of AL is more aggressive, with a median survival of < 6 months in untreated symptomatic patients, compared to 2-6 years in ATTR-CA [72].
The faster progression of AL amyloidosis may result from the proven cytotoxic effects of human amyloidogenic lightchain oligomers. Accumulation of these oligomers in the cardiac muscle leads to impaired cardiomyocyte contractility through oxidative stress. This has been confirmed in a study comparing AL-CA, ATTRwt-CA and ATTRv-CA, where despite lower amyloid accumulation in the LV wall measured using LGE in AL patients, similar decreases in LS and overall greater impairment of LV function were observed compared to patients with higher LGE in the TTR mutation groups. Early identification of patients with AL amyloidosis is also essential for initiating treatment, which is more readily available and can significantly help a large proportion of patients, unlike in the case of ATTR-CA, where drugs are still in the research phase and the therapeutic path is not as clearly defined [73].
Currently, a diagnostic protocol based on single-photon emission computed tomography (SPECT) with both 99mTcPYP and 99mTc-DPD is used for non-invasive differentiation of these two diseases. This imaging has a 99% negative predictive value and 86% positive predictive value. The lower specificity was associated with slight tracer uptake in individuals with AL, hence a combination of this method with immunofixation of monoclonal protein in blood or urine was proposed, raising the specificity to 100%. Additionally, a 4-point scale based on visual marker assessment in bone, heart, and soft tissues has been introduced to facilitate the description of the SPECT imaging [74].
Conventional histopathology and amyloid typing are still necessary in cases of uncertainty (e.g., suspicion of MGUS or chronic kidney disease coexisting with ATTR-CA, overlapping characteristic clinical features of both CA subtypes) despite the methods used or their inaccessibility. The highest sensitivity is seen with kidney or liver biopsies (90%). Other less invasive procedures perform worse, for example biopsies of skin (70-80%), abdominal fat tissue (60-80%), rectum (50-70%) or bone marrow (50-60%). The biopsy of abdominal fat tissue is often preferred for screening due to its high availability and ease of procedure when more accurate methods are not accessible. Endomyocardial biopsy, which carries greater risks, is reserved for heart-related angiography or suspected cases of isolated CA. If an endomyocardial biopsy does not reveal the characteristic microscopic pattern, it can help rule out heart involvement in amyloidosis. In that cases TTR amyloidosis is diagnosed by identifying a TTR gene mutation, along with Congo red staining and anti-TTR antibody labeling of an endomyocardial or extracardiac biopsy sample. Whereas, diagnosis of AL amyloidosis involves detecting high levels of monoclonal protein in serum and/or urine, along with Congo red staining and labeling with specific anti-k or anti-l light-chain antibodies on an endomyocardial or extracardiac biopsy. TTRwt amyloidosis is diagnosed when an endomyocardial biopsy shows both red Congo staining and labeling with anti-TTR antibodies, or positive staining on an extracardiac biopsy associated with significant cardiac uptake of technetium 99 bisphosphonate during scintigraphy in the absence of any TTR mutation [75].
Possibilities of rapid diagnosis of asymptomatic patients
As for the timing of screening, it is recommended to start screening 10 years before the estimated age of onset of symptoms (the estimate takes into account the typical age of onset of ATTR-CA and the age of diagnosis of ATTR-CA in the family) [76]. ATTR-CA is the only form of CA in which the diagnosis can be made non-invasively [8]. At this point, the most sensitive screening test is cardiac scintigraphy (SPECT) with tracers such as 99mTechnetium-pyrophosphate (99mTc-PYP), 99mTechnetium-3,3-diphosphono-1,2-propanodicarboxylic (99mTcDPD), 99mTechnetium hydroxymethylene diphosphonate (99mTc-HMDP), in which tracer uptake has been observed in asymptomatic patients [77-78]. The method has proven sensitive and has made it possible to distinguish AL-CA from ATTR-CA, as uptake is almost non-existent in the former [39], although it is not completely specific for ATTR-CA [79]. According to the algorithm for non-invasive diagnosis of amyloidosis, the absence of clonal proliferation along with high 99mTc-PYP/DPD/HMDP uptake in SPECT suggests ATTR-CA with high probability, without the need for endomyocardial biopsy [80].
Another imaging study to identify asymptomatic patients is CMR with LGE, which can show characteristic deposits even before the thickening of the ventricular wall. Combining this method with T1-mapping imaging [81] and extracellular volume measurements can identify patients at risk of developing ATTR-CA with 80-93% sensitivity and specificity [82-83].
The role of genetic testing in identifying pre-penetration carriers of TTR variants cannot be overlooked [84]. If detection of the variant itself by mass spectroscopy is not sufficient (or the result is negative), and there is a high clinical suspicion of ATTR-CA, then deoxyribonucleic acid (DNA) sequencing is used. Sometimes the sequence of actions taken before the biopsy can be reversed depending on availability of diagnostic techniques and clinical case [4].
Uncertainties about conventional treatment
Another challenge is effective treatment to both prevent the development of HF and improve the condition of patients with ATTR-developed heart failure. Little is known about the benefits of conventional HF therapy in patients with ATTR-CA and most recommendations are based on expert opinion. Many reports focus on the increased risk of hypotonia in patients with ATTR-CA due to the reduced ability to maintain stroke volume by increasing the volume and force of ventricular contractility. Therefore, these patients’ cardiac output largely dependent on heart rate and caution is advised when administering beta blockers which are indicated only when atrial arrhythmias coexist and at the lowest possible dose [41].
Also, the use of angiotensin-converting-enzyme inhibitors, angiotensin receptor blockers, angiotensin receptor/ neprilysin inhibitors and sodium-glucose co-transporter-2 (SGLT2) inhibitors raises concerns about symptomatic hypotension, particularly with autonomic neuropathy co-occurring with ATTR-CA [85-86]. A solution in such cases may be the use of midodrine or droxidopa and compression stockings [87].
Although the use of loop diuretics is the mainstay of maintaining fluid balance in patients with ATTR-CA and their intravenous use reduces hospital and emergency department admissions [88], one should avoid aggressively forcing diuresis, as there is a high risk of organ hypoperfusion and consequent acute kidney injury [85]. The first-line drug is usually furosemide, whereas torasemide or bumetanide is recommended if there is no response. Synergistic aldosterone antagonists or thiazides may be added as a next step [6]. There is an increased need for diuretics as HF progresses and the need for high doses is an unfavorable prognostic factor [89]. Similarly, the use of vasopressors has been associated with a particularly high mortality rate, which may be due to the cardiogenic shock these patients are usually admitted with [90].
Because of the difficulty in establishing adequate fluid balance, the narrow therapeutic window, and the increased risk of hypotony and hypoperfusion in patients with ATTR-CA treated with standard HF medications, a strategy based on remote monitoring might be beneficial in this group [91]. In light of recent reports and recommendations on the treatment of HF [92], it seems important to further investigate the effect of SGLT2-inhibitors on the course of ATTR-CA.
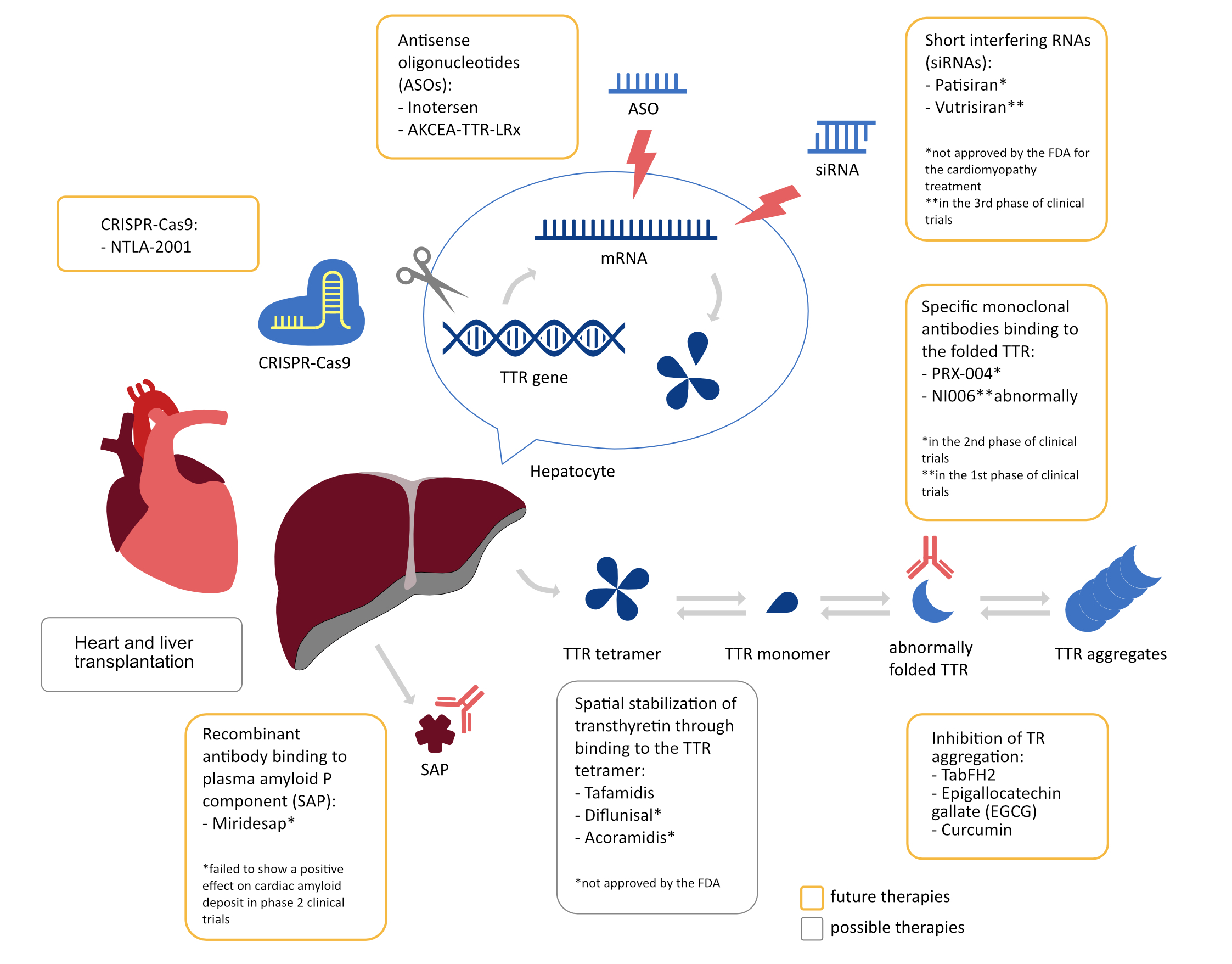
Future therapies
CRISPR-Cas9 is a genetic engineering method consisting of two components. First, guideRNA recognizes a target sequence in the cell’s genome and guides the Cas9 enzyme to it, which activates the “cutting” process. A defective DNA fragment with a pathogenic mutation can be “cut out” or a repair-like process can be induced by homologous recombination based on the provided RNA template. NTLA-2001 is a drug using this mechanism and it caused a > 97% decrease in TTR in mice after a single application over 12 months [93].
Another strategy is to degrade or extract amyloid using specific monoclonal antibodies. PRX-004 is an antibody that binds only to abnormally folded TTR, thus it may improve the macrophagal removal of amyloid accumulated in the myocardium [94]. In a phase I study (NCT03336580), PRX-004 shows efficacy and a good safety profile. A phase II trial (NCT05442047) is currently underway to focus specifically on patients with ATTR-CA, and the results of this trial are to be published by May 2025 [95]. Another antibody in phase I clinical trials with a similar mechanism is NI006, which, after 12 months of us, caused a decrease in radio tracer uptake in cardiac scintigraphy and extracellular volume in CMR [96].
A type of recombinant antibody targeting the plasma amyloid P component (SAP) is Miridesap. SAP is synthesized in the liver and binds to all types of amyloid fibers and administration of Miridesap was associated with a > 90% decrease in serum SAP and activation of complement-dependent phagocytosis of amyloid deposits [97]. It also showed a reduction of the amyloid deposit burden in liver and kidneys over 6 weeks of therapy, but patients with cardiac involvement were not included in the study [98]. A subsequent phase 2 clinical trial failed to show a positive effect on cardiac amyloid deposits with ATTR-CA, and further studies of Miridesap were abandoned [99].
If there is a high load of amyloid deposits, the process of deposition of native TTR in tissues and organs on preformed amyloid fibrils can continue despite its stabilization. In such a case, the protein association inhibitor TabFH2, which binds to the F- and H- ends of beta-strands and blocks the process of TTR deposition, may be helpful [100]. There are also other inhibitors of TTR aggregation – a study of 19 patients with ATTR-CA taking epigallocatechin gallate (EGCG), a polyphenol that in vitro shows the ability to inhibit the formation of amyloid deposits [101], for 12 months showed a 12.5% decrease in mean LV mass, a 9% increase in mean mitral annular velocity and no disease progression [102]. However, a later retrospective study on a larger group found no effect of EGCG on mortality [103]. Also, curcumin administered chronically to mice with the human TTR p.V50M variant slowed tetrameric dissociation and reduced deposition of amyloid deposits, moreover, it seems to be able to dissolve the deposits [104]. Unfortunately, it is poorly tolerated at high doses, has a short half-life and poor bioavailability [6], hence the need for its pharmacokinetic modification if it were to be applicable to this indication. The available and currently investigated treatment methods are summarized in Figure 4.
Figure 4. Summary of ATTR treatment methods and their mechanisms of action
Conclusions
Amyloidosis includes diseases like AL and ATTR, with an increasing diagnosis of ATTR cardiomyopathy (ATTR-CA). ATTR is divided into wild-type (ATTRwt) and variant (ATTRv), with p.V142I being common. Genetic screening is crucial due to higher mortality and severe symptoms in ATTRv. ATTR-CA results in restrictive cardiomyopathy and diastolic heart failure, leading to complications like atrial fibrillation, bradyarrhythmias, and occasionally ventricular tachycardias, necessitating interventions such as pacemakers, anticoagulation, and ICDs. Diagnosing ATTR-CA involves echocardiography, cardiac scintigraphy and CMR with LGE. Differentiating it from AL amyloidosis is challenging, requiring comprehensive diagnostics. Treatments like liver transplantation are beneficial, especially for Val30Met mutation patients. Disease-modifying therapies, including Tafamidis, antisense oligonucleotides and siRNAs, show promise. Future treatments explore CRISPR-Cas9, monoclonal antibodies, protein association inhibitors and natural compounds like EGCG and curcumin.
Acknowledgment
None.
Conflict of interest
None.
Funding
None.
References
| 1. |
Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol [Internet]. 2015;24(6):343–50. Available from: https://linkinghub.elsevier.com/retrieve/pii/S105488071500085X.
|
| 2. |
ESC Cardiomyopathy and Myocarditis Registry [Internet]. [cited 2023 Oct 11]. Available from: https://www.escardio.org/Research/registries.
|
| 3. |
Rasmussen TB, Ladefoged BT, Dybro AM, Clemmensen TS, Sørensen RH, Terkelsen AJ, et al. Transthyretin Gene Variants and Associated Phenotypes in Danish Patients with Amyloid Cardiomyopathy. Cardiogenetics [Internet]. 2022;12(1):1–11. Available from: https://www.mdpi.com/2035-8148/12/1/1.
|
| 4. |
Migliaccio MG, Iodice F, Di Mauro M, Iannuzzi A, Pacileo R, Caiazza M, et al. Cardiac Amyloidosis: Diagnostic Tools for a Challenging Disease. Cardiogenetics [Internet]. 2021;11(3):111–21. Available from: https://www.mdpi.com/2035-8148/11/3/12.
|
| 5. |
Dubrey S. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM [Internet]. 1998;91(2):141–57. Available from: https://academic.oup.com/qjmed/article-lookup/doi/10.1093/qjmed/91.2.141.
|
| 6. |
Griffin JM, Rosenthal JL, Grodin JL, Maurer MS, Grogan M, Cheng RK. ATTR Amyloidosis: Current and Emerging Management Strategies. JACC CardioOncology [Internet]. 2021;3(4):488–505. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2666087321001551.
|
| 7. |
Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, et al. Misdiagnosis of Hereditary Amyloidosis as AL (Primary) Amyloidosis. N Engl J Med [Internet]. 2002;346(23):1786–91. Available from: http://www.nejm.org/doi/abs/10.1056/NEJMoa013354.
|
| 8. |
Ihne S, Morbach C, Sommer C, Geier A, Knop S, Störk S. Amyloidosis — the Diagnosis and Treatment of an Underdiagnosed Disease. Dtsch Arztebl Int [Internet]. 2020; Available from: https://www.aerzteblatt.de/10.3238/arztebl.2020.0159.
|
| 9. |
Adams D, Ando Y, Beirão JM, Coelho T, Gertz MA, Gillmore JD, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol [Internet]. 2021;268(6):2109–22. Available from: https://link.springer.com/10.1007/s00415-019-09688-0.
|
| 10. |
Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, et al. Variant-Sequence Transthyretin (Isoleucine 122) in Late-Onset Cardiac Amyloidosis in Black Americans. N Engl J Med [Internet]. 1997;336(7):466–73. Available from: http://www.nejm.org/doi/abs/10.1056/NEJM199702133360703.
|
| 11. |
Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The Amyloidogenic V122I Transthyretin Variant in Elderly Black Americans. N Engl J Med [Internet]. 2015;372(1):21–9. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1404852.
|
| 12. |
Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis. J Am Coll Cardiol [Internet]. 2016;68(2):161–72. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109716330108.
|
| 13. |
Akinboboye O, Shah K, Warner AL, Damy T, Taylor HA, Gollob J, et al. DISCOVERY: prevalence of transthyretin ( TTR ) mutations in a US-centric patient population suspected of having cardiac amyloidosis. Amyloid [Internet]. 2020;27(4):223–30. Available from: https://www.tandfonline.com/doi/full/10.1080/13506129.2020.1764928.
|
| 14. |
Rowczenio D, Quarta CC, Fontana M, Whelan CJ, Martinez-Naharro A, Trojer H, et al. Analysis of the TTR gene in the investigation of amyloidosis: A 25-year single UK center experience. Hum Mutat [Internet]. 2019;40(1):90–6. Available from: https://onlinelibrary.wiley.com/doi/10.1002/humu.23669.
|
| 15. |
Iodice F, Di Mauro M, Migliaccio MG, Iannuzzi A, Pacileo R, Caiazza M, et al. Cardiac Amyloidosis Therapy: A Systematic Review. Cardiogenetics [Internet]. 2021;11(1):10–7. Available from: https://www.mdpi.com/2035-8148/11/1/2.
|
| 16. |
Gillmore JD, Damy T, Fontana M, Hutchinson M, Lachmann HJ, Martinez-Naharro A, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J [Internet]. 2018;39(30):2799–806. Available from: https://academic.oup.com/eurheartj/article/39/30/2799/4557556.
|
| 17. |
Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, Fontana M, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Hear Fail [Internet]. 2019;12(9). Available from: https://www.ahajournals.org/doi/10.1161/CIRCHEARTFAILURE.119.006075.
|
| 18. |
Maurizi N, Rella V, Fumagalli C, Salerno S, Castelletti S, Dagradi F, et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int J Cardiol [Internet]. 2020;300:191–5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0167527319309350.
|
| 19. |
Nitsche C, Scully PR, Patel KP, Kammerlander AA, Koschutnik M, Dona C, et al. Prevalence and Outcomes of Concomitant Aortic Stenosis and Cardiac Amyloidosis. J Am Coll Cardiol [Internet]. 2021;77(2):128–39. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109720377354.
|
| 20. |
Sekijima Y, Uchiyama S, Tojo K, Sano K, Shimizu Y, Imaeda T, et al. High prevalence of wild-type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol [Internet]. 2011;42(11):1785–91. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0046817711001444.
|
| 21. |
Bhuiyan T, Helmke S, Patel AR, Ruberg FL, Packman J, Cheung K, et al. Pressure-Volume Relationships in Patients With Transthyretin (ATTR) Cardiac Amyloidosis Secondary to V122I Mutations and Wild-Type Transthyretin. Circ Hear Fail [Internet]. 2011;4(2):121–8. Available from: https://www.ahajournals.org/doi/10.1161/CIRCHEARTFAILURE.109.910455.
|
| 22. |
Chacko L, Martone R, Bandera F, Lane T, Martinez-Naharro A, Boldrini M, et al. Echocardiographic phenotype and prognosis in transthyretin cardiac amyloidosis. Eur Heart J [Internet]. 2020;41(14):1439–47. Available from: https://academic.oup.com/eurheartj/article/41/14/1439/5708960.
|
| 23. |
Sartiani L, Bucciantini M, Spinelli V, Leri M, Natalello A, Nosi D, et al. Biochemical and Electrophysiological Modification of Amyloid Transthyretin on Cardiomyocytes. Biophys J [Internet]. 2016;111(9):2024–38. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0006349516308037.
|
| 24. |
Giancaterino S, Urey MA, Darden D, Hsu JC. Management of Arrhythmias in Cardiac Amyloidosis. JACC Clin Electrophysiol [Internet]. 2020;6(4):351–61. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2405500X20300797.
|
| 25. |
El-Am EA, Dispenzieri A, Melduni RM, Ammash NM, White RD, Hodge DO, et al. Direct Current Cardioversion of Atrial Arrhythmias in Adults With Cardiac Amyloidosis. J Am Coll Cardiol [Internet]. 2019;73(5):589–97. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109718393914.
|
| 26. |
Donnellan E, Elshazly MB, Vakamudi S, Wazni OM, Cohen JA, Kanj M, et al. No Association Between CHADS-VASc Score and Left Atrial Appendage Thrombus in Patients With Transthyretin Amyloidosis. JACC Clin Electrophysiol [Internet]. 2019;5(12):1473–4. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2405500X1930831X.
|
| 27. |
Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, et al. Intracardiac Thrombosis and Embolism in Patients With Cardiac Amyloidosis. Circulation [Internet]. 2007;116(21):2420–6. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.107.697763.
|
| 28. |
Mints YY, Doros G, Berk JL, Connors LH, Ruberg FL. Features of atrial fibrillation in wild‐type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC Hear Fail [Internet]. 2018;5(5):772–9. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ehf2.12308.
|
| 29. |
Donnellan E, Wazni O, Kanj M, Elshazly MB, Hussein A, Baranowski B, et al. Atrial fibrillation ablation in patients with transthyretin cardiac amyloidosis. EP Eur [Internet]. 2020;22(2):259–64. Available from: https://academic.oup.com/europace/article/22/2/259/5625716.
|
| 30. |
Donnellan E, Wazni OM, Hanna M, Elshazly MB, Puri R, Saliba W, et al. Atrial Fibrillation in Transthyretin Cardiac Amyloidosis. JACC Clin Electrophysiol [Internet]. 2020;6(9):1118–27. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2405500X20303376.
|
| 31. |
Sanchis K, Cariou E, Colombat M, Ribes D, Huart A, Cintas P, et al. Atrial fibrillation and subtype of atrial fibrillation in cardiac amyloidosis: clinical and echocardiographic features, impact on mortality. Amyloid [Internet]. 2019;26(3):128–38. Available from: https://www.tandfonline.com/doi/full/10.1080/13506129.2019.1620724.
|
| 32. |
Fernandes A, Caetano F, Almeida I, Paiva L, Gomes P, Mota P, et al. Diagnostic approach to cardiac amyloidosis: A case report. Rev Port Cardiol (English Ed [Internet]. 2016;35(5):305.e1-305.e7. Available from: https://linkinghub.elsevier.com/retrieve/pii/S217420491630023X.
|
| 33. |
Algalarrondo V, Dinanian S, Juin C, Chemla D, Bennani SL, Sebag C, et al. Prophylactic pacemaker implantation in familial amyloid polyneuropathy. Hear Rhythm [Internet]. 2012;9(7):1069–75. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1547527112002019.
|
| 34. |
Glikson M, Nielsen JC, Kronborg MB, Michowitz Y, Auricchio A, Barbash IM, et al. 2021 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy. Eur Heart J [Internet]. 2021;42(35):3427–520. Available from: https://academic.oup.com/eurheartj/article/42/35/3427/6358547.
|
| 35. |
Donnellan E, Wazni OM, Hanna M, Kanj M, Saliba WI, Jaber WA. Cardiac Resynchronization Therapy for Transthyretin Cardiac Amyloidosis. J Am Heart Assoc [Internet]. 2020;9(14). Available from: https://www.ahajournals.org/doi/10.1161/JAHA.120.017335.
|
| 36. |
Kristen A V., Dengler TJ, Hegenbart U, Schonland SO, Goldschmidt H, Sack F-U, et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Hear Rhythm [Internet]. 2008;5(2):235–40. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1547527107010260.
|
| 37. |
Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J [Internet]. 2015;36(41):2793–867. Available from: https://academic.oup.com/eurheartj/article-lookup/doi/10.1093/eurheartj/ehv316.
|
| 38. |
Sławiński G, Dorniak K, Lewicka E. Implantable cardioverter-defibrillators in cardiac amyloidosis: a grey zone requiring an individual approach. Kardiol Pol [Internet]. 2020;78(9):934–6. Available from: https://journals.viamedica.pl/kardiologia_polska/article/view/82656.
|
| 39. |
Scully PR, Patel KP, Treibel TA, Thornton GD, Hughes RK, Chadalavada S, et al. Prevalence and outcome of dual aortic stenosis and cardiac amyloid pathology in patients referred for transcatheter aortic valve implantation. Eur Heart J [Internet]. 2020;41(29):2759–67. Available from: https://academic.oup.com/eurheartj/article/41/29/2759/5817909.
|
| 40. |
Arana-Achaga X, Goena-Vives C, Villanueva-Benito I, Solla-Ruiz I, Rengel Jimenez A, Gaspar TI, et al. Development and Validation of a Prediction Model and Score for Transthyretin Cardiac Amyloidosis Diagnosis. JACC Cardiovasc Imaging [Internet]. 2023;16(12):1567–80. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1936878X23002218.
|
| 41. |
Rubin J, Maurer MS. Cardiac Amyloidosis: Overlooked, Underappreciated, and Treatable. Annu Rev Med [Internet]. 2020;71(1):203–19. Available from: https://www.annualreviews.org/doi/10.1146/annurev-med-052918-020140.
|
| 42. |
Xu B, Godoy Rivas C, Rodriguez ER, Tan C, Gillinov AM, Harb S, et al. Unrecognized Cardiac Amyloidosis at the Time of Mitral Valve Surgery: Incidence and Outcomes. Cardiology [Internet]. 2019;142(4):253–8. Available from: https://karger.com/CRD/article/doi/10.1159/000499933.
|
| 43. |
Volz MJ, Pleger ST, Weber A, Geis NA, Hamed S, Mereles D, et al. Initial experience with percutaneous mitral valve repair in patients with cardiac amyloidosis. Eur J Clin Invest [Internet]. 2021;51(6). Available from: https://onlinelibrary.wiley.com/doi/10.1111/eci.13473.
|
| 44. |
Giannini F, Colombo A. Percutaneous treatment of tricuspid valve in refractory right heart failure. Eur Hear J Suppl [Internet]. 2019;21(Supplement_B):B43–7. Available from: https://academic.oup.com/eurheartjsupp/article/21/Supplement_B/B43/5422944.
|
| 45. |
Holmgren G, Steen L, Suhr O, Ericzon B-G, Groth C-G, Andersen O, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet [Internet]. 1993;341(8853):1113–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/014067369393127M.
|
| 46. |
Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology [Internet]. 2010;75(4):324–7. Available from: https://www.neurology.org/doi/10.1212/WNL.0b013e3181ea15d4.
|
| 47. |
Kazi DS, Bellows BK, Baron SJ, Shen C, Cohen DJ, Spertus JA, et al. Cost-Effectiveness of Tafamidis Therapy for Transthyretin Amyloid Cardiomyopathy. Circulation [Internet]. 2020;141(15):1214–24. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.119.045093.
|
| 48. |
Lohrmann G, Pipilas A, Mussinelli R, Gopal DM, Berk JL, Connors LH, et al. Stabilization of Cardiac Function With Diflunisal in Transthyretin (ATTR) Cardiac Amyloidosis. J Card Fail [Internet]. 2020;26(9):753–9. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1071916419314356.
|
| 49. |
Stoilova T, Colombo L, Forloni G, Tagliavini F, Salmona M. A New Face for Old Antibiotics: Tetracyclines in Treatment of Amyloidoses. J Med Chem [Internet]. 2013;56(15):5987–6006. Available from: https://pubs.acs.org/doi/10.1021/jm400161p.
|
| 50. |
Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med [Internet]. 2018;379(11):1007–16. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1805689.
|
| 51. |
Hammarström P, Schneider F, Kelly JW. Trans -Suppression of Misfolding in an Amyloid Disease. Science (80- ) [Internet]. 2001;293(5539):2459–62. Available from: https://www.science.org/doi/10.1126/science.1062245.
|
| 52. |
Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci [Internet]. 2013;110(24):9992–7. Available from: https://pnas.org/doi/full/10.1073/pnas.1300761110.
|
| 53. |
Crooke ST, Witztum JL, Bennett CF, Baker BF. RNA-Targeted Therapeutics. Cell Metab [Internet]. 2018;27(4):714–39. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1550413118301827.
|
| 54. |
Dasgupta NR, Rissing SM, Smith J, Jung J, Benson MD. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid [Internet]. 2020;27(1):52–8. Available from: https://www.tandfonline.com/doi/full/10.1080/13506129.2019.1685487.
|
| 55. |
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med [Internet]. 2018;379(1):22–31. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1716793.
|
| 56. |
Viney NJ, Guo S, Tai L, Baker BF, Aghajan M, Jung SW, et al. Ligand conjugated antisense oligonucleotide for the treatment of transthyretin amyloidosis: preclinical and phase 1 data. ESC Hear Fail [Internet]. 2021;8(1):652–61. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ehf2.13154.
|
| 57. |
Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis [Internet]. 2015;10(1):109. Available from: http://www.ojrd.com/content/10/1/109.
|
| 58. |
Ioannou A, Fontana M, Gillmore JD. Patisiran for the Treatment of Transthyretin-mediated Amyloidosis with Cardiomyopathy. Heart Int [Internet]. 2023;17(1):27. Available from: https://www.touchcardio.com/cardiovascular-disease/journal-articles/patisiran-for-the-treatment-of-transthyretin-mediated-amyloidosis-with-cardiomyopathy/.
|
| 59. |
Solomon SD, Adams D, Kristen A, Grogan M, González-Duarte A, Maurer MS, et al. Effects of Patisiran, an RNA Interference Therapeutic, on Cardiac Parameters in Patients With Hereditary Transthyretin-Mediated Amyloidosis. Circulation [Internet]. 2019;139(4):431–43. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.118.035831.
|
| 60. |
McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J [Internet]. 2021;42(36):3599–726. Available from: https://academic.oup.com/eurheartj/article/42/36/3599/6358045.
|
| 61. |
Rosenblum H, Masri A, Narotsky DL, Goldsmith J, Hamid N, Hahn RT, et al. Unveiling outcomes in coexisting severe aortic stenosis and transthyretin cardiac amyloidosis. Eur J Heart Fail [Internet]. 2021;23(2):250–8. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ejhf.1974.
|
| 62. |
Ikeda S, Sekijima Y, Tojo K, Koyama J. Diagnostic value of abdominal wall fat pad biopsy in senile systemic amyloidosis. Amyloid [Internet]. 2011;18(4):211–5. Available from: http://www.tandfonline.com/doi/full/10.3109/13506129.2011.623199.
|
| 63. |
González-López E, Gagliardi C, Dominguez F, Quarta CC, de Haro-del Moral FJ, Milandri A, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J [Internet]. 2017;38(24):1895–904. Available from: https://academic.oup.com/eurheartj/article/38/24/1895/3059372.
|
| 64. |
Carroll JD, Gaasch WH, McAdam KPWJ. Amyloid cardiomyopathy: Characterization by a distinctive voltage/mass relation. Am J Cardiol [Internet]. 1982;49(1):9–13. Available from: https://linkinghub.elsevier.com/retrieve/pii/0002914982902703.
|
| 65. |
Dorbala S, Ando Y, Bokhari S, Dispenzieri A, Falk RH, Ferrari VA, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2—evidence base and standardized methods of imaging. J Nucl Cardiol [Internet]. 2019;26(6):2065–123. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1071358123063900.
|
| 66. |
Cueto-Garcia L, Reeder GS, Kyle RA, Wood DL, Seward JB, Naessens J, et al. Echocardiographic findings in systemic amyloidosis: Spectrum of cardiac involvement and relation to survival. J Am Coll Cardiol [Internet]. 1985;6(4):737–43. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109785804757.
|
| 67. |
Phelan D, Collier P, Thavendiranathan P, Popović ZB, Hanna M, Plana JC, et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart [Internet]. 2012;98(19):1442–8. Available from: https://heart.bmj.com/lookup/doi/10.1136/heartjnl-2012-302353.
|
| 68. |
Fontana M, Pica S, Reant P, Abdel-Gadir A, Treibel TA, Banypersad SM, et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation [Internet]. 2015;132(16):1570–9. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.115.016567.
|
| 69. |
Holcman K, Kostkiewicz M, Podolec P, Rubiś P. Cardiac amyloidosis—state-of-the-art diagnosis and emerging therapies [in Polish]. Folia Cardiol [Internet]. 2019;14(6):625–33. Available from: https://journals.viamedica.pl/folia_cardiologica/article/view/FC.2019.0115.
|
| 70. |
Hanson JLS, Arvanitis M, Koch CM, Berk JL, Ruberg FL, Prokaeva T, et al. Use of Serum Transthyretin as a Prognostic Indicator and Predictor of Outcome in Cardiac Amyloid Disease Associated With Wild-Type Transthyretin. Circ Hear Fail [Internet]. 2018;11(2). Available from: https://www.ahajournals.org/doi/10.1161/CIRCHEARTFAILURE.117.004000.
|
| 71. |
Kapoor M, Foiani M, Heslegrave A, Zetterberg H, Lunn MP, Malaspina A, et al. Plasma neurofilament light chain concentration is increased and correlates with the severity of neuropathy in hereditary transthyretin amyloidosis. J Peripher Nerv Syst [Internet]. 2019;24(4):314–9. Available from: https://onlinelibrary.wiley.com/doi/10.1111/jns.12350.
|
| 72. |
Holcman K, Kostkiewicz M, Podolec P, Rubiś P. Amyloidoza serca — właściwe rozpoznanie i nowe terapie na horyzoncie [in Polish]. Folia Cardiol [Internet]. 2019;14(6):625–33. Available from: https://journals.viamedica.pl/folia_cardiologica/article/view/67034.
|
| 73. |
Falk RH. Pondering the Prognosis and Pathology of Cardiac Amyloidosis. JACC Cardiovasc Imaging [Internet]. 2016;9(2):139–41. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1936878X15008633.
|
| 74. |
Perugini E, Guidalotti PL, Salvi F, Cooke RMT, Pettinato C, Riva L, et al. Noninvasive Etiologic Diagnosis of Cardiac Amyloidosis Using 99m Tc-3,3-Diphosphono-1,2-Propanodicarboxylic Acid Scintigraphy. J Am Coll Cardiol [Internet]. 2005;46(6):1076–84. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109705014075.
|
| 75. |
Żelichowski G. Diagnostyka amyloidozy układu sercowo-naczyniowego [in Polish]. Chor Serca i Naczyń [Internet]. 2010;7(1):40–8. Available from: https://journals.viamedica.pl/choroby_serca_i_naczyn/article/view/12024/9902.
|
| 76. |
Conceição I, Damy T, Romero M, Galán L, Attarian S, Luigetti M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations*. Amyloid [Internet]. 2019;26(1):3–9. Available from: https://www.tandfonline.com/doi/full/10.1080/13506129.2018.1556156.
|
| 77. |
Haq M, Pawar S, Berk JL, Miller EJ, Ruberg FL. Can 99mTc-Pyrophosphate Aid in Early Detection of Cardiac Involvement in Asymptomatic Variant TTR Amyloidosis? JACC Cardiovasc Imaging [Internet]. 2017;10(6):713–4. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1936878X16304430.
|
| 78. |
Minutoli F, Di Bella G, Mazzeo A, Laudicella R, Gentile L, Russo M, et al. Serial scanning with 99mTc-3, 3-diphosphono-1, 2-propanodicarboxylic acid (99mTc-DPD) for early detection of cardiac amyloid deposition and prediction of clinical worsening in subjects carrying a transthyretin gene mutation. J Nucl Cardiol [Internet]. 2021;28(5):1949–57. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1071358123015507.
|
| 79. |
Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in Cardiac Amyloidosis: A Review. J Am Heart Assoc [Internet]. 2012;1(2). Available from: https://www.ahajournals.org/doi/10.1161/JAHA.111.000364.
|
| 80. |
Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation [Internet]. 2016;133(24):2404–12. Available from: https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.116.021612.
|
| 81. |
Vogelsberg H, Mahrholdt H, Deluigi CC, Yilmaz A, Kispert EM, Greulich S, et al. Cardiovascular Magnetic Resonance in Clinically Suspected Cardiac Amyloidosis. J Am Coll Cardiol [Internet]. 2008;51(10):1022–30. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109707038727.
|
| 82. |
Pan JA, Kerwin MJ, Salerno M. Native T1 Mapping, Extracellular Volume Mapping, and Late Gadolinium Enhancement in Cardiac Amyloidosis. JACC Cardiovasc Imaging [Internet]. 2020;13(6):1299–310. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1936878X20303041.
|
| 83. |
White JA, Kim HW, Shah D, Fine N, Kim K-Y, Wendell DC, et al. CMR Imaging With Rapid Visual T1 Assessment Predicts Mortality in Patients Suspected of Cardiac Amyloidosis. JACC Cardiovasc Imaging [Internet]. 2014;7(2):143–56. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1936878X13008152.
|
| 84. |
Damrauer SM, Chaudhary K, Cho JH, Liang LW, Argulian E, Chan L, et al. Association of the V122I Hereditary Transthyretin Amyloidosis Genetic Variant With Heart Failure Among Individuals of African or Hispanic/Latino Ancestry. JAMA [Internet]. 2019 Dec 10;322(22):2191. Available from: https://jamanetwork.com/journals/jama/fullarticle/2757227.
|
| 85. |
Kittleson MM, Maurer MS, Ambardekar A V., Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation [Internet]. 2020 Jul 7;142(1). Available from: https://www.ahajournals.org/doi/10.1161/CIR.0000000000000792.
|
| 86. |
González-Duarte A, Barroso F, Mundayat R, Shapiro B. Blood pressure and orthostatic hypotension as measures of autonomic dysfunction in patients from the transthyretin amyloidosis outcomes survey (THAOS). Auton Neurosci [Internet]. 2019;222:102590. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1566070219301043.
|
| 87. |
Palma J-A, Gonzalez-Duarte A, Kaufmann H. Orthostatic hypotension in hereditary transthyretin amyloidosis: epidemiology, diagnosis and management. Clin Auton Res [Internet]. 2019;29(S1):33–44. Available from: http://link.springer.com/10.1007/s10286-019-00623-x.
|
| 88. |
Vaishnav J, Hubbard A, Chasler JE, Lepley D, Cuomo K, Riley S, et al. Management of heart failure in cardiac amyloidosis using an ambulatory diuresis clinic. Am Heart J [Internet]. 2021;233:122–31. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0002870320304117.
|
| 89. |
Cheng RK, Levy WC, Vasbinder A, Teruya S, De Los Santos J, Leedy D, et al. Diuretic Dose and NYHA Functional Class Are Independent Predictors of Mortality in Patients With Transthyretin Cardiac Amyloidosis. JACC CardioOncology [Internet]. 2020;2(3):414–24. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2666087320301563.
|
| 90. |
d’Humières T, Fard D, Damy T, Roubille F, Galat A, Doan H-L, et al. Outcome of patients with cardiac amyloidosis admitted to an intensive care unit for acute heart failure. Arch Cardiovasc Dis [Internet]. 2018;111(10):582–90. Available from: https://linkinghub.elsevier.com/retrieve/pii/S187521361830055X.
|
| 91. |
Abraham WT, Adamson PB, Bourge RC, Aaron MF, Costanzo MR, Stevenson LW, et al. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet [Internet]. 2011;377(9766):658–66. Available from: https://linkinghub.elsevier.com/retrieve/pii/S187521361830055X.
|
| 92. |
McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J [Internet]. 2023;44(37):3627–39. Available from: https://academic.oup.com/eurheartj/article/44/37/3627/7246292.
|
| 93. |
Finn JD, Smith AR, Patel MC, Shaw L, Youniss MR, van Heteren J, et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep [Internet]. 2018;22(9):2227–35. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2211124718301827.
|
| 94. |
George J, Rappaport M, Shimoni S, Goland S, Voldarsky I, Fabricant Y, et al. A novel monoclonal antibody targeting aggregated transthyretin facilitates its removal and functional recovery in an experimental model. Eur Heart J [Internet]. 2020;41(12):1260–70. Available from: https://academic.oup.com/eurheartj/article/41/12/1260/5684841.
|
| 95. |
PRX004, the First Investigational Anti-Amyloid Immunotherapy for the Treatment of ATTR Amyloidosis. 9-month results from a Phase 1 long-term extension study [Internet]. 2020. Available from: https://s201.q4cdn.com/351053094/files/doc_presentations/2020/12/1/PRX004-Phase-1-results-(PDF)-12.8.2020[3].pdf.
|
| 96. |
Garcia-Pavia P, aus dem Siepen F, Donal E, Lairez O, van der Meer P, Kristen A V., et al. Phase 1 Trial of Antibody NI006 for Depletion of Cardiac Transthyretin Amyloid. N Engl J Med [Internet]. 2023 Jul 20;389(3):239–50. Available from: http://www.nejm.org/doi/10.1056/NEJMoa2303765.
|
| 97. |
Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature [Internet]. 2010;468(7320):93–7. Available from: https://www.nature.com/articles/nature09494.
|
| 98. |
Richards DB, Cookson LM, Berges AC, Barton S V., Lane T, Ritter JM, et al. Therapeutic Clearance of Amyloid by Antibodies to Serum Amyloid P Component. N Engl J Med [Internet]. 2015;373(12):1106–14. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1504942.
|
| 99. |
Richards DB, Cookson LM, Barton S V., Liefaard L, Lane T, Hutt DF, et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci Transl Med [Internet]. 2018;10(422). Available from: https://www.science.org/doi/10.1126/scitranslmed.aan3128.
|
| 100. |
Saelices L, Nguyen BA, Chung K, Wang Y, Ortega A, Lee JH, et al. A pair of peptides inhibits seeding of the hormone transporter transthyretin into amyloid fibrils. J Biol Chem [Internet]. 2019;294(15):6130–41. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0021925820366825.
|
| 101. |
Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol [Internet]. 2008;15(6):558–66. Available from: https://www.nature.com/articles/nsmb.1437.
|
| 102. |
Kristen A V., Lehrke S, Buss S, Mereles D, Steen H, Ehlermann P, et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin Res Cardiol [Internet]. 2012;101(10):805–13. Available from: http://link.springer.com/10.1007/s00392-012-0463-z.
|
| 103. |
Cappelli F, Martone R, Taborchi G, Morini S, Bartolini S, Angelotti P, et al. Epigallocatechin-3-gallate tolerability and impact on survival in a cohort of patients with transthyretin-related cardiac amyloidosis. A single-center retrospective study. Intern Emerg Med [Internet]. 2018;13(6):873–80. Available from: http://link.springer.com/10.1007/s11739-018-1887-x.
|
| 104. |
Ferreira N, Saraiva M, Almeida M. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int J Mol Sci [Internet]. 2019;20(6):1287. Available from: https://www.mdpi.com/1422-0067/20/6/1287.
|
