
Pediatric cardiology emergencies – the role of Long QT Syndrome among arrhythmias


Abstract
Long QT syndrome (LQTS) is a hereditary disease with significant mortality, which might be reduced with appropriate management. This cardiac disorder is regarded as rare, but its prevalence remains unknown. The clinical course of LQTS is variable and syncope is a common first manifestation of LQTS. Therefore in each patient after syncope an ECG should be carried out. However, there is no universal QT value applying to all patients (especially infants and children), because it varies depending on age and sex. Genetic testing can be of great importance for the management of families with LQTS and early identification of patient relatives at risk of developing disease. We aimed to show that a very important part of treatment is not only pharmacotherapy, especially beta-blockers, but change of lifestyle plays a significant role.
Citation
Zipser M, Zienciuk-Krajka A, Krenska D, Kwiatkowska J. Pediatric cardiology emergencies – the role of Long QT Syndrome among arrhythmias. Eur J Transl Clin Med. 2018;1(2):4-10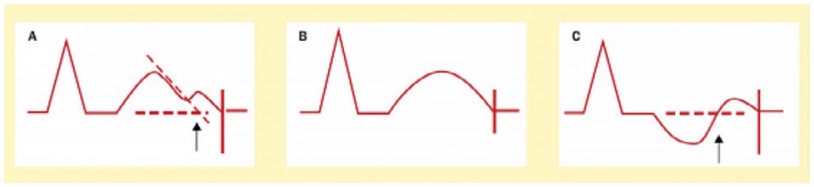






Introduction
Syncope is a common complaint of children and young adults visiting emergency departments. In terms of etiology, syncope is classified as reflex syncope (including vasovagal), syncope due to orthostatic hypotension and cardiac syncope [1].
Arrhythmias such as bradyarrhythmias or tachyarrhythmias are the most common cardiogenic causes of syncope. The former includes hemodynamically significant bradycardia in e.g. advanced atrio-ventricular block or Sick Sinus Syndrome. Whereas the latter lead to syncope due to reduced ejection fraction because of fast, hemodynamically insufficient ventricular contractions. Another reason of cardiogenic syncope is Long QT Syndrome (LQTS) with high risk of fatal ventricular arrhythmias such as ventricular fibrillation (VF) and torsade de pointes (TdP). However, LQTS is treatable and therefore it needs to be excluded early in the diagnostic process of patients with recurring syncope or with family history of sudden cardiac death (particularly of an infant or a child).
In this article we describe the pathomechanism and clinical picture of LQTS as well as the diagnostic pitfalls associated with it.
Definition and genetic basis
LQTS is a hereditary arrhythmia syndrome caused by mutations of genes coding for ion channels and their accessory proteins in cardiomyocytes. Abnormal ion transport prolongs the repolarization of cardiac muscle, thus increasing the risk of fatal ventricular arrhythmias (VF and TdP). At rest, the patient's ECG might reveal a prolonged QT interval, abnormal T waves and a tendency for bradycardia [2]. Therefore, a more precise name would be "prolonged QT and abnormal T-wave syndrome" [3]. It is proven that the T-wave patterns in ECGs of LQTS patients not only present genotype dependence, but also vary dynamically on exercise stress test or due to epinephrine, b-blockers or glucose.
Clinically, LQTS may present as syncope, tremor or sudden cardiac death, particularly in situations with increased sympathetic nervous system activity (stress, physical effort, intense emotions). Mutations of 19 different genes were linked with sodium/potassium channel dysfunction that prolongs cardiomyocyte repolarization [4]. Three of them are the basis of ~90% of all genetically confirmed cases of LQTS: LQT1 (50%), LQT2 (35%) and LQT3 (8%). Despite the advanced diagnostic methods, 15-20% of patients with long QT interval have no abnormalities in their genetic tests [5].
Epidemiology
The incidence of LQTS varies according to the source of information. A prospective analysis of the QT interval in ECGs revealed LQTS in 1 out of 2500 Italian newborns, which is more frequent than previously reported [4]. It is noteworthy that about 20-25% of patients with genetically confirmed LQTS have a normal ECG tracing at rest, thus all studies based on ECG analysis underestimate its incidence. Autopsy studies reveal mutations of genes responsible for sodium/ potassium channels in 5-10% of patients who died of Sudden Infant Death Syndrome (SIDS).
Secondary LQTS
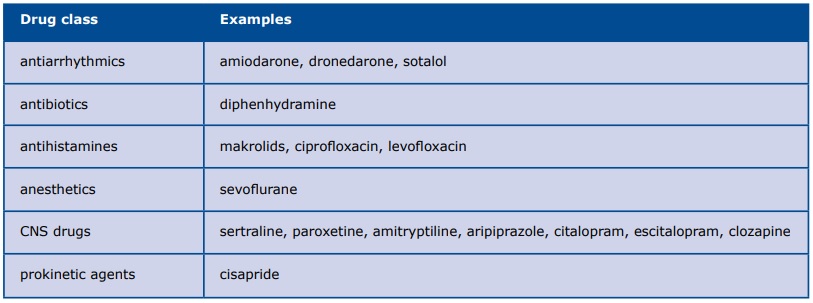
Although LQTS by definition is caused by a gene mutation, clinicians should not forget about the secondary causes of LQTS which are treatable: severe bradycardia, electrolyte deficiencies (hypokalemia, hypomagnesemia, hypocalcemia) and drugs (anesthetics, antiarrhythmics, antibiotics, antihistamines, CNS drugs and prokinetic agents) (Table 1). For more details see www.crediblemeds.org.
Table 1. Drugs causing secondary LQTS
Amiodarone deserves attention due to its wide and often long-term use as the antiarrhythmic drug. Despite causing a significant prolongation of QT interval, amiodarone only slightly increases the risk of TdP, due to its ability to inhibit early after depolarizations (EADs). The occurrence of TdP in patients on long-term amiodarone therapy is probably due to an electrolyte deficiency or concurrent treatment with other QT-prolonging drugs [6]. Regardless, patients with long-term amiodarone therapy they all should have their QT interval measured at every follow-up visit.
Clinical picture of LQTS
LQTS is often asymptomatic and the first clinical manifestation can be cardiac arrest due to TdP progressing to VF. Symptoms of LQTS can be either clinical (due to experiencing an episode of arrhythmia) or electrocardiographic (based on ECG at rest).
Patients with LQTS most often experience episodes of TdP, a polymorphic ventricular tachycardia named by French cardiologist Dessertenne in 1966 after the alternating electric axis of the ventricular complexes. TdP typically involves short episodes of tachycardia 200-250/min with either spontaneous return of sinus rhythm or progression to VF. During a TdP episode, the work of the ventricles is hemodynamically insufficient and the intensity of symptoms is related to the duration of the episode. Patients with short episodes of TdP might complain of palpitations, dizziness and weakness. Longer episodes may cause syncope, convulsions and cardiac arrest. There are many factors known to provoke episodes of arrhythmia, however typical for LQTS are situations in which loss of consciousness (TdP) provokes: physical exertion, emotional stress and sleep. Of note is the increased risk of sudden cardiac death during swimming and exposure to loud sounds. Symptoms might appear as early as the first months of life, with peak at 10-30 years of age [7].
Diagnostic tests
The first and critical step in diagnosing LQTS is taking a detailed history from the patient. The physician must focus on the history of episodes of pre/syncope and their circumstances (was there a transient loss of consciousness? how quick was the recovery? any concurrent injuries or convulsions?). The physician should ask detailed questions in order to confirm or rule out vasovagal syncope (most frequent cause of syncope in children and young adults). Another critical subject is family history of sudden and/or unexplained death at a young age (car accidents, drownings, miscarriages).
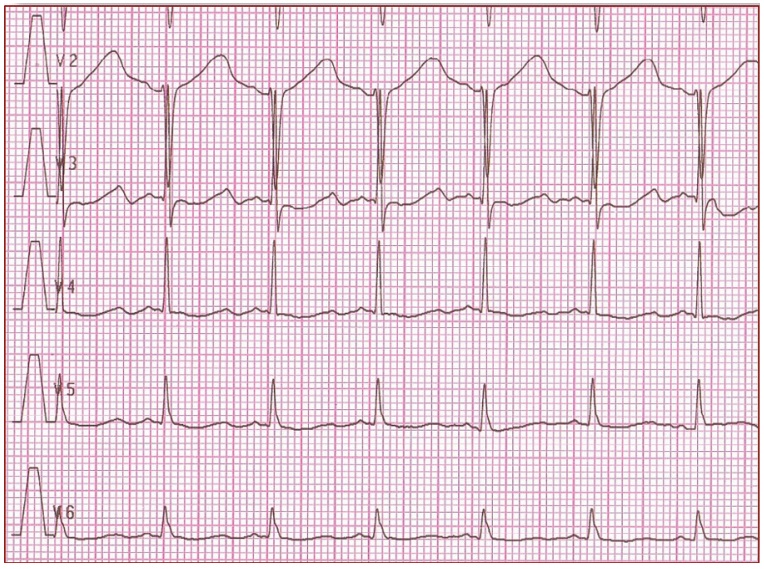
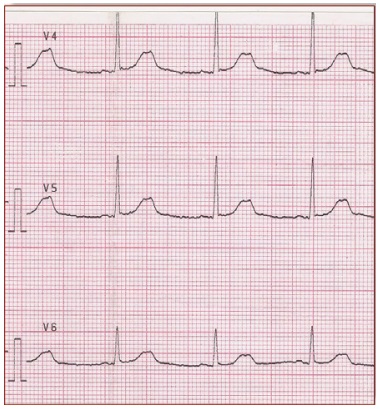
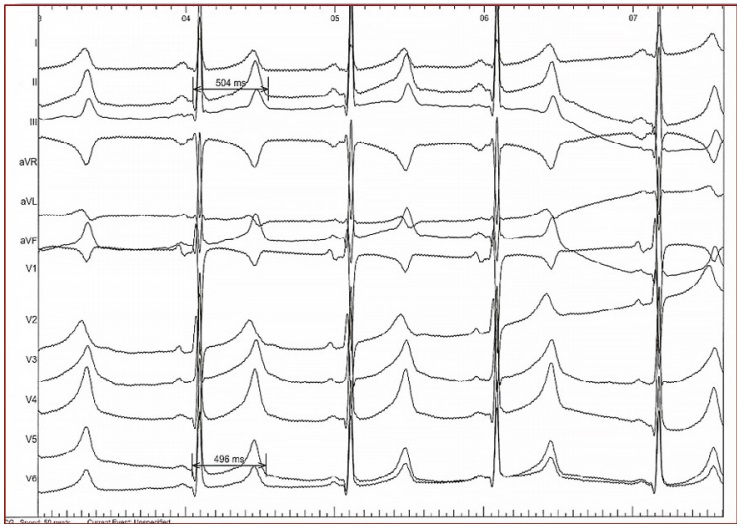
In daily practice, the cornerstone of LQTS diagnosis continues to be the QT and QTc assessment in a 12- lead ECG taken at rest (Figure 1) [8-11]. ECG tracings of LQTS patients at rest may reveal a rather wide spectrum of changes in the heart rate and repolarization phase (Figure 2-4: LQT 1-3 types). Specific changes in the repolarization phase are as follows: prolonged QT interval, abnormal T waves (wide, notched, low or high voltage) or alternating T waves (indicating significant electrical instability of the heart) (Figure 5). The repolarization phase (QT interval measured from the beginning of Q wave to the end of the T wave) depends on the patient's heart rate, age and sex. Therefore, we rely on the so-called corrected QT interval (QTc) calculated using Bazett's formula and it should be used optimally for heart rate between 60-120(100)/min: QTc=QT/(√RR).
Figure 1. Diagram shows the QT interval measurements rules [2]
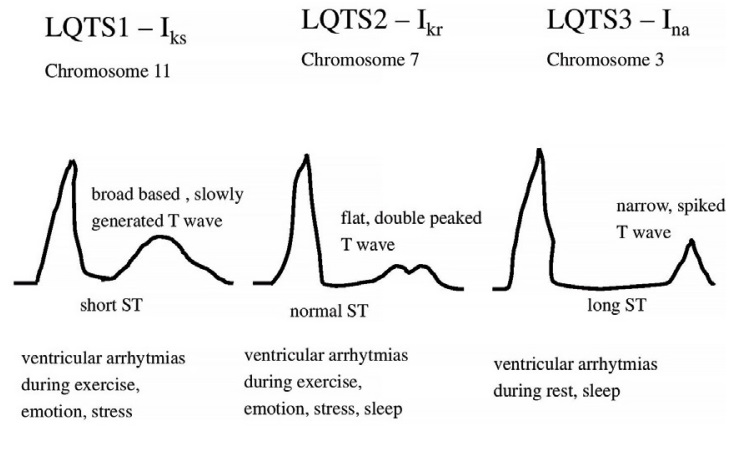
Figure 2. It shows 'early onset' broad based, slowly generated T waves. LQT1 (R591H)
Figure 3. It shows small late T waves. Sometimes these T waves will be notched or double peaked in lead V4. LQT2 (G601S)
Figure 4. It shows a flat and prolonged ST segment with a 'late onset' normal-shaped T wave. LQT3 (K1477N)
Figure 5. Abnormal types of T waves in LQTS (LQTS1, LQTS2, LQTS3)
Newborns and infants with QTc of 440-470ms in repeated ECG need further diagnostics especially taking detailed family history including LQTS and SCD, Holter ECG. For older children of both sexes, QTc of 450ms is currently considered normal and is slightly affected by growing. QTc >460ms for adult females and 450ms for adult males should be investigated. In case of noting QTc >500ms and excluding possible secondary causes, a diagnosis of LQTS can be made. In case of borderline QTc values (460-480ms), it is necessary to take into account the entire clinical picture and to assess the probability of LQTS based on Schwartz’s diagnostic criteria.
The Schwartz diagnostic criteria published in 1993 continue to be the tool of choice for stratifying the risk of LQTS. Included are clinical symptoms, family history and ECG changes. According to current guidelines, LQTS should be recognised in case of >3 points in Schwartz scale [12].
Multicenter studies revealed correlations between the LQT genotype and the specific clinical picture of LQTS (arrhythmia-provoking factors, benefits of beta- -blocker therapy and age groups at greatest risk of SCD) [13]. Such correlations highlight the usefulness of genetic testing, even in cases when there are doubts about the clinical diagnosis. Confirming the patient's precise genotype allows a more precise therapy e.g. adding sodium channel blockers in therapy of patients with LQT3 genotype [12]. On the other hand patients with LQT2 are more sensitive to hypokalemia. In addition, genetic testing allows making the diagnosis in the asymptomatic family members of LQTS patients.
Due to the incomplete penetrance of LQTS genes, some of the patients with genetically confirmed LQTS actually have a normal QT interval in resting ECG. Diagnosing this subset of patients is particularly challenging because they are clinically and electrocardiographically asymptomatic. Often these patients have family members who either survived cardiac arrest or have definite ECG changes. These patients must avoid QT-prolonging drugs. Other tests such as Holter ECG, exercise stress test and provocative tests play less important role [14].
The next step in diagnostic process should be stratification for the risk of Sudden Cardiac Death. Traditional SCD risk factors are as follows: aborted cardiac arrest (ACA) at < 1 year of age, prior syncope, torsade de Pointes, T-wave alternans, QTc>500ms, males <14 years old, females 18-40 years old, congenital deafness (JLN).
Treatment
The actual recommendations for LQTS management include genetic-specific treatment and lifestyle advice [15]. Specifically, this means chronic treatment with propranolol or mexiletine (currently not sold in Poland and available only upon special request for import), avoidance of QT-prolonging drugs, electrolyte/ hydration replenishment, aggressive fever reduction, genetic testing whenever possible, access to AED, BLS training for the entire family, psychological support and family screening.
During an episode of TdP, the management is identical regardless if the etiology is primary/genetic LQTS or secondary. The drug of choice is magnesium sulphate administered intravenously either in fast boluses children: 3-12mg/kg in 1-2 minutes; adults: 1-2g in 30- 60 seconds) or continuous infusion (children: 0.5-1mg/ kg/h; adults: 3-10mg/min). The suggested magnesium sulphate concentration in pediatric dosing is 3-5mg/ dl [16]. Electric cardioversion should be reserved for the hemodynamically unstable patients because TdP frequently recurrrent after electric discharges. Next steps in management should focus on treating the cause, if possible (e.g. correcting the electrolyte imbalance, discontinuing the QT-prolonging drug).
Beta-blockers propranolol and nadolol have been the mainstay of LQTS treatment for years. They are able to prevent syncope and SCD in up to 70% of LQTS patients [15]. The particularly poor effectiveness of beta-blockers is noted in patients with LQT3 genotype, in whom ventricular tachycardias occur during sleep or at rest. Most authors point to propranolol (3mg/kg 3 times/day in children) and nadolol (1mg/kg 1-2 times daily). Atenolol should be avoided [17]. Despite their lower effectiveness, beta-blockers are also recommended in LQT3 therapy.
In case of lack of response to drug treatment or cardiac arrest, the next step is implantation of a cardioverter-defibrillator or left stellate ganglioectomy (LSG) [18]. Side effects of LSG are: intermittent temperature changes, facial flush, decreased sweating on one side and pain.
The literature describes numerous cases of LQTS patients misdiagnosed with seizure disorders. This is particularly common among patients whose symptoms include muscle tremors or recurrent syncope. We suggest all patients treated for recurring seizures should have an ECG performed before increasing doses or adding another drug for epilepsy resistant to treatment. MacCormick et al. noted that a delay in diagnosis of LQTS “for those who were misdiagnosed as having epilepsy, the median time for diagnostic delay was 11.8 years, with a range of 9.5 to 23 years” [19]. Considering the high mortality due to LQTS (20% in the first year and 50% within 5 years after an episode of transient unconsciousness), there is little time for making the correct diagnosis [19-20]. It is worth underlining that symptomatic LQTS is the only opportunity for excluding it in his/her closest family members, therefore lack of correct diagnosis is not just one patient’s risk.
Conclusions
Long QT syndrome is a rare cardiac cause of transient loss of consciousness. However, its high mortality and high effectiveness of preventive treatment means that every patient presenting with syncope or convulsions should have an assessment of the QT interval and T waves on a 12-lead ECG. This is especially true of patients with a family history of sudden or unexplained death. Due to an increasing number of prescription drugs which prolong the repolarization phase in cardiac muscle (thus increasing the risk of TdP), physicians of all specialties should be trained in QT assessment and when in doubt should have an easy access to a cardiology consultation. It appears that information about the patient’s QT interval is just as important in the planning of treatment, as information allergies and adverse reactions to drugs. Some authors suggest that an ECG screening program for newborns is justified, as it would reduce the frequency of sudden death in the entire pediatric population and would at the same time meet the criteria of cost-effectiveness accepted in European healthcare systems [21].
References
| 1. |
Brignole M, Moya A, De Lange FJ, Deharo J-C, Elliott PM, Fanciulli A, et al. 2018 ESC Guidelines for the diagnosis and management of syncope. Kardiol Pol. 2018;76(8):1119-98.
|
| 2. |
Lanjewar P, Pathak V, Lokhandwala Y. Issues in QT interval measurement. Indian Pacing Electrophysiol J. 2004;4(4):156-61.
|
| 3. |
Horie M. Long QT syndrome presents not only as QT prolongation but also as abnormal T-wave morphology. Heart Rhythm. 2017;14(8):1171-2.
|
| 4. |
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120(18):1761-7.
|
| 5. |
Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011;13(8):1077-109.
|
| 6. |
Antonelli D, Atar S, Freedberg NA, Rosenfeld T. Torsade de pointes in patients on chronic amiodarone treatment: contributing factors and drug interactions. IMAJ. 2005;7:163-5.
|
| 7. |
Rohatgi RK, Sugrue A, Bos JM, Cannon BC, Asirvatham SJ, Moir C, et al. Contemporary outcomes in patients with long QT syndrome. J Am Coll Cardiol. 2017;70(4):453-62.
|
| 8. |
Baranowski R, Bieganowska K, Kozłowski D, Kukla P, Kurpesa M, Lelakowski J, et al. Zalecenia dotyczące stosowania rozpoznań elektrokardiograficznych. Kardiol Pol. 2010;68(supl IV):335-89.
|
| 9. |
Rijnbeek PR, Witsenburg M, Schrama E, Hess J, Kors JA. New normal limits for the paediatric electrocardiogram. Eur Heart J. 2001;22(8):702-11.
|
| 10. |
Schwartz PJ, Garson A, Paul T, Stramba-Badiale M, Vetter VL, Villain E, et al. Guidelines for the interpretation of the neonatal electrocardiogram: a task force of the European Society of Cardiology. Eur Heart J. 2002;23(17):1329-44.
|
| 11. |
Viskin S, Rosovski U, Sands AJ, Chen E, Kistler PM, Kalman JM, et al. Inaccurate electrocardiographic interpretation of long QT: the majority of physicians cannot recognize a long QT when they see one. Heart Rhythm. 2005;2(6):569-74.
|
| 12. |
Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Hear Rhythm. 2018;15(10):e73-189.
|
| 13. |
Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89-95.
|
| 14. |
Obeyesekere MN, Klein GJ, Modi S, Leong-Sit P, Gula LJ, Yee R, et al. How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythmia Electrophysiol. 2011;4(6):958-64.
|
| 15. |
Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2015 Nov 1;36(41):2793-867.
|
| 16. |
Hoshino K, Ogawa K, Hishitani T, Isobe T, Etoh Y. Successful uses of magnesium sulfate for torsades de pointes in children with long QT syndrome. Pediatr Int. 2006;48(2):112-7.
|
| 17. |
Kwok S, Pflaumer A, Pantaleo S, Date E, Jadhav M, Davis AM. Ten‐year experience in atenolol use and exercise evaluation in children with genetically proven long QT syndrome. J arrhythmia. 2017;33(6):624-9.
|
| 18. |
Surman TL, Stuklis RG, Chan JC. Thoracoscopic Sympathectomy for Long QT Syndrome. Literature Review and Case Study. Heart Lung Circ. 2018 Feb;In Press, Corrected Proof.
|
| 19. |
MacCormick JM, McAlister H, Crawford J, French JK, Crozier I, Shelling AN, et al. Misdiagnosis of long QT syndrome as epilepsy at first presentation. Ann Emerg Med. 2009;54(1):26-32.
|
| 20. |
Rola R, Ryglewicz D. Neuronal channelopathies. Postępy Nauk Med. 2012;25(1):51-9.
|
| 21. |
Quaglini S, Rognoni C, Spazzolini C, Priori SG, Mannarino S, Schwartz PJ. Cost-effectiveness of neonatal ECG screening for the long QT syndrome. Eur Heart J. 2006;27(15):1824-32.
|
