
Rare histological subtypes of renal cell carcinoma in everyday diagnostic practice


Abstract
The fourth edition of the WHO Classification of Tumours of the Urinary System and Male Genital Organs (2016) contains new renal tumour entities. These new subtypes of renal cell carcinoma (RCC) were introduced based on morphological criteria, some genetic features, and clinical characteristics with prognostic implications.
We present three patients with rare renal tumours belonging to newly recognized or still emerging categories of RCC. All cases were diagnosed based on careful morphological examination with immunophenotyping, and patho- clinical correlation. The first case is an example of acquired cystic disease - associated renal cell carcinoma with heterogeneous architecture as well as specific intra- and intercytoplasmic microlumens. The second tumour – a tubulocystic renal cell carcinoma – was composed of multiple, various-sized cysts divided by fibrovascular septa and tubules lined focally with hobnail cells. The third case presents very rare sporadic eosinophilic, solid, and cystic RCC. This tumour contained macro et microcystic, areas intermixed with solid fields composed of large, in part multinucleated eosinophilic cells. Inflammatory infiltrations accompanied the neoplastic stroma. New subtypes of RCC, although rare, can be encountered in everyday practice. It is important to perform careful differential diagnosis and classify such tumours according to the recent guidelines.
Citation
Hypszer M, Czarnota K, Kobierska-Gulida G, Nałęcz A, Wesołowski W, Iżycka-Świeszewska E. Rare histological subtypes of renal cell carcinoma in everyday diagnostic practice. Eur J Transl Clin Med. 2018;1(2):64-71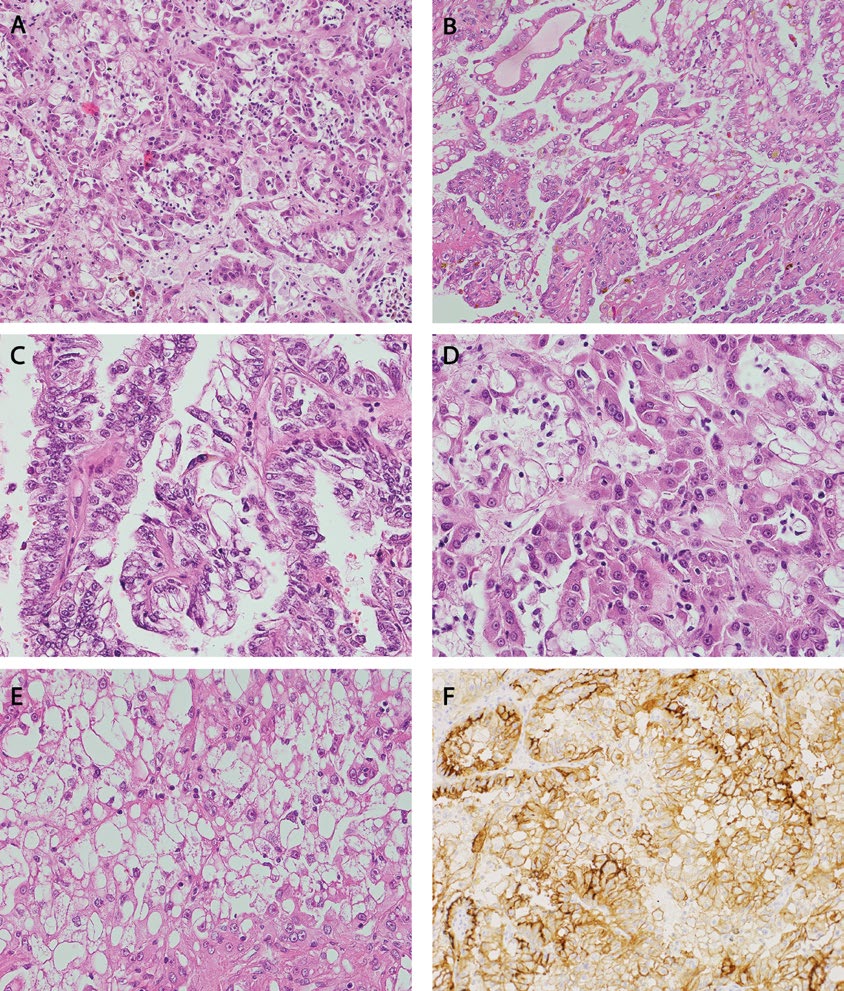



Introduction
The classification of renal neoplasia is still based on morphology but it has been evolving dynamically over the past years due to advances in the understanding of molecular pathogenesis of these tumours [1]. Correct diagnosis of renal tumours carries significant clinical implications for patients such as prognostic risk stratification, selection of targeted therapeutics and identification of cases for further genetic testing [1-3].
The last WHO Classification of Tumours of the Urinary System and Male Genital Organs (2016) recognizes several distinct RCC subtypes. New tumour entities include: hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cell carcinoma, Succinate dehydrogenase-deficient renal cell carcinoma, tubulocystic renal cell carcinoma, acquired cystic disease-associated renal cell carcinoma, and clear cell papillary renal cell carcinoma [1, 4-5]. In addition, the classification recognizes also tumour types described as emerging, which have distinct histological and genetic pattern but due to their rarity there is yet not enough data to include them as separate subtypes [1, 4-5].
The incidence and mortality due to renal cell carcinomas (RCC) have been increasing over the last years and it the 9th most common cancer in men and 14th in women worldwide [5]. The majority of cases occur in countries with high socioeconomic status [5]. The established risk factors for RCC are cigarette smoking, obesity and certain occupational exposures [3]. Hypertension or its treatment, especially using diuretics has also been associated with increased risk of these tumours [3, 5]. The incidence of renal cell cancer is increased 3-6 times in patients with acquired cystic kidney disease [3, 5]. Most renal cell carcinomas are sporadic but 2-4 % have familial causes [3]. The risk of renal cancer for a first degree relative of a patient with renal cancer is double [5]. There are several genetic disorders associated with RCC such as von Hippel-Lindau syndrome (clear cell RCC), hereditary papillary RCC, hereditary leiomyomatosis and RCC, familial papillary thyroid carcinoma (papillary RCC), hyperparathyroidism- jaw tumour syndrome, Birt-Hogg-Dube syndrome, tuberous sclerosis, and constitutional chromosome 3 translocations [3, 5].
In this article we present three cases of renal cell carcinomas, that disclose distinct histological and clinical features, and belong either to newly recognized or emerging entities.
Material and methods
Three out of 47 cases of renal tumours diagnosed in routine practice in 2017 in the El-Pat Laboratory of Pathology and the Department of Pathomorphology at the Copernicus Hospital. The cases included in the article were the ones that needed consultation and second opinion. The clinical data included patients’ age, sex, basic medical history, radiological imaging results.
All tumours underwent routine pathological procedures- gross examination with adequate tissue sampling and histological examination with wide panel immunophenotyping. Immunohistochemistry was performed based on DAKO auto-stainer antibodies against: CK AE 1/3, CK 7, CK 8/18, CK 19, CK 20, CD10, CD117, AMACR, CA IX, EMA, S100, PAX 8, inhibin, estrogen receptor, vimentin and Ki67; with appropriate producer recommendations.
Literature review was performed in PubMed; the references used in the article were published in the years 2006-2019.
Results
Patient 1
A 58-year old man was admitted to the hospital for a scheduled dialysis. For 6 years he has been suffering from end-stage renal disease caused by poorly managed type 2 diabetes. The patient was asymptomatic. A routine abdominal ultrasound examination revealed a mass in the left kidney. CT scan confirmed a hypodense tumour in the superior pole of the kidney, 6.0 x 5.0 x 4.5 cm. The cortex of both kidneys was thin, and there was also a 1.5 cm cyst in the cortex of the left kidney. The patient underwent radical left nephrectomy.
On macroscopic examination, the kidney measured 12 x 7 x 7 cm. Underneath the capsule of the superior pole, there was a well-circumscribed soft tumour (5.5 cm in its greatest diameter) with a grey cut surface. The tumour didn't break the renal capsule and didn't invade the renal pelvis (pT1b) [6].
Histological examination showed well-circumscribed neoplasm with heterogeneous architecture, mainly composed of solid and papillary areas as well as cribriform/sieve-like pattern (Figure 1A, B, C). The neoplastic cells were large, partially columnar, with eosinophilic granular cytoplasm showing intracytoplasmatic and intercytoplasmatic microlumens (Fig. 1D, E) with rare oxalate crystals – clear to slightly opaque, polarizable structures seen in intracytoplasmic spaces. The nuclei were vesicular with variably conspicuous nucleoli (International Society of Urological Pathology (ISUP) scale grade 2-4, and overall showed intermediate atypia. Deposits of hemosiderin were present as well as foci of chronic inflammatory infiltrate. The neoplastic cells showed immunopositivity for CD10 (Figure 1F), AMACR, vimentin and were negative for CK7, CD117 and CAIX. Proliferation index measured with Ki67 was around 2%. The kidney tissue surrounding tumour presented chronic inflammatory changes with vascular changes, parenchymal fibrosis, as well as small cysts deriving from renal tubules. Based on tumour morphology, renal pathology, and supplied patient clinical history, acquired cystic disease (ACD)-associated renal cell carcinoma was diagnosed.
Figure 1 A-F.
Microscopic findings in Acquired cystic disease-RCC: A – Various architectural patterns (solid, tubular and papillary) (HE, 200x); B – Solid and cystic architectural patterns (HE, 200x); C – Papillary and macrocystic architectural patterns (HE, 400x); D, E – Distinctive intra – and intercytoplasmic microlumens; nucleoli are very prominent (HE, 400x); F – immunoreactivity for CD10 (200x)
Patient 2
A 65-year old woman was incidentally diagnosed with a 2 cm mass in the left kidney, found on CT-scan and confirmed on MRI examination. In the next weeks, the patient underwent partial nephrectomy.
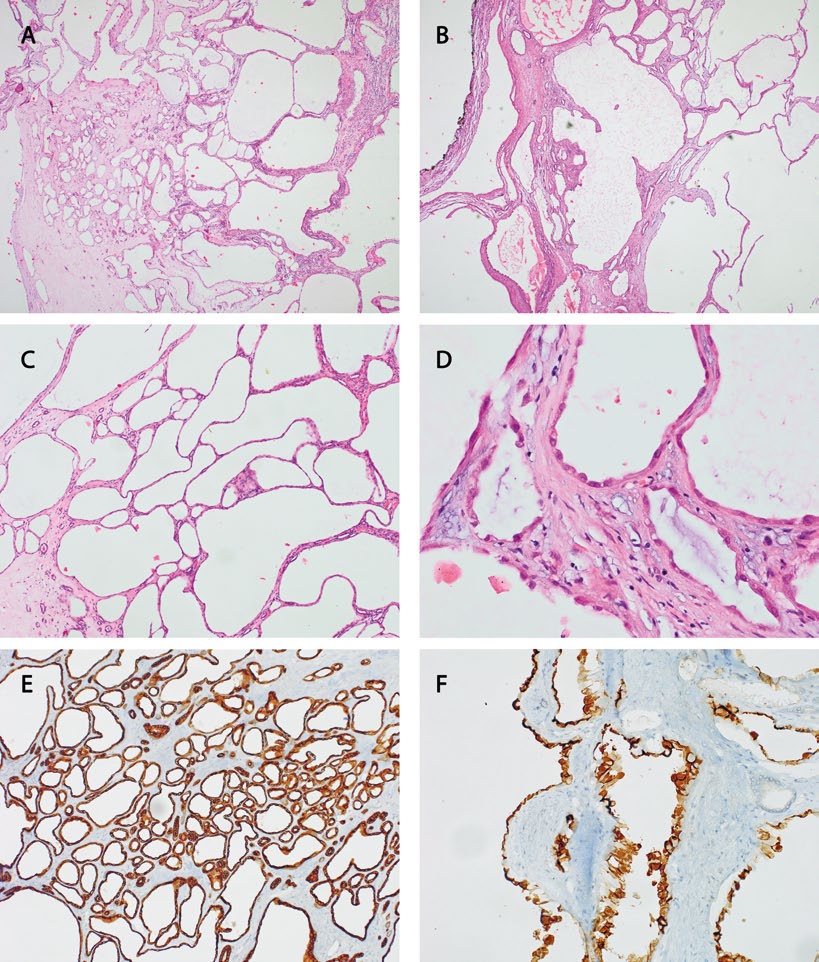
Gross examination showed a multicystic tumour measuring 2.2 x 1.5 x 2.2 cm (pT1a) [6]. On microscopic examination the tumour was composed of tubules (Figure 2A) and multiple cysts of various size (Figure 2B, C), focally lined with cells with distinctive hobnail pattern (Figure 2D); other cells were flattened. The cysts were separated by fibrovascular septa. The cytoplasm of neoplastic cells was eosinophilic, nuclei were round with mild atypia, some had visible nucleoli (ISUP grade 2, singular cells with ISUP grade 3 nucleoli). Cells were immunopositive for AMACR, CD10, vimentin, CAIX (weak staining), CK8/18, CK19 (Figure 2F) and focally for CK7 (Figure 2E), while negative for inhibin and estrogen receptors. Proliferation Ki67 index was <1%.
The tumour was diagnosed as tubulocystic renal cell carcinoma.
Figure 2 A-F.
Microscopic findings in Tubulocystic-RCC: A – Predominately micro- and macrocystic architectural patterns (HE, 40x); B –
Outermost part of the tumour (HE, 40x); C – Cystic part of the tumour (HE, 100x); D – Flattened, cuboidal and hobnail cells lining tumour
cysts (HE, 400x); E – CK7 highlighting tubular part of the tumour (100x); F – CK19 immunoreactivity prominent in the hobnail cells lining the
lumens (200x)
Patient 3
A 77-year old woman with history of arterial hypertension complained of abdominal pain. The ultrasound revealed a pathological mass up to 2.5 cm in the upper pole of her left kidney. A CT scan confirmed a tumour in the upper half of the left kidney, with no clear margins, without signs of breaking renal capsule or involving renal pelvis. The patient underwent partial nephrectomy.
On macroscopic examination, the tumour measured 2.0 x 2.5 x 2.0 cm and was partially solid with cystic spaces, white-grey on cut surface with small red spots (pT1a).
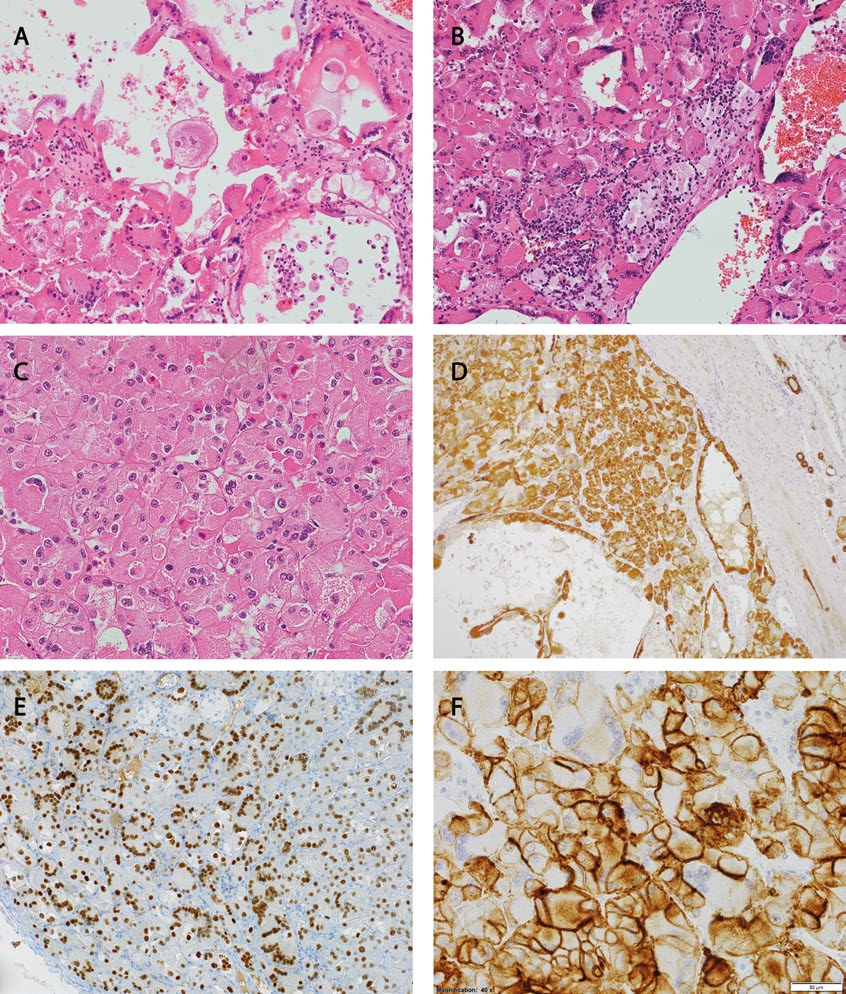
Histologically, macrocystic spaces were lined with cells with eosinophilic cytoplasm (Figure 3A) and solid parts (Figure 3 B, C) were composed of large round cells with abundant eosinophilic cytoplasm, hyperchromatic nuclei with mild nuclear pleomorphism and discrete nucleoli (ISUP grade 2). In addition, there was an admixture of lymphocytes and histiocytes and multinucleated giant eosinophilic cells within the solid areas. The neoplastic cells were positive for AMACR (patchy cytoplasmic staining), vimentin, CKAE1/AE3 (Figure 3D), PAX8 (strong nuclear reaction, Figure 3E), CD10 (Figure 3F), CK20 staining showed focal positivity. In parallel, the tumour was negative for CK7, CD117, EMA, S-100, CAIX. Proliferation Ki67 index was 2%. Based on careful differential diagnosis, and after literature review the tumour was finally diagnosed as a renal cell carcinoma, subtype eosinophilic solid and cystic [7].
Figure 3 A-F.
Microscopic findings in Eosinophilic, solid and cystic-RCC: A – More cystic part of the tumour, note bright pink cytoplasm and the
various shape of cells lining the cysts (HE, 200x); B – Predominately solid part of the tumour, with multiple giant cells, lymphocytes, and histiocytes (HE, 200x); C – Solid part of the tumour, cells are bright pink with easily noticeable nucleoli (HE, 400x); D – Positive staining for CK AE1/3; also highlighting pre-existing tubules in the healthy part of the kidney on the right side of the photo (100x); E – Nuclear expression of PAX8, note the highlighted giant-cells nuclei (200x); F – Strong membranous staining for CD10; here the multinucleated giant cells are negative (400x)
Discussion
In 2016 the 4th edition of the WHO Classification of Tumours of the Urinary System and Male Genital Organs introduced important changes into histological approach to renal tumours [5, 8]. As mentioned before, genetics play an increased role in new classifications challenging and sometimes changing our understanding of tumour pathology. While clear cell carcinoma and papillary carcinoma remain the most common renal cancers, careful examination combined with exact clinical data can lead to an exact diagnosis of even rare subtypes and therefore better clinical management. Furthermore, the latest WHO classification recommends using the ISUP scale for tumour grading based on nucleoli appearance (grade 1-3) and cell pleomorphism (grade 4) instead of the traditional Fuhrman scale [5]. The latter, due to its methodology problems (e.g. an overly complicated grading based on 3 seperate parameters, which can cause discordance, imprecise criteria regarding nuclear pleomorphism) and unsatisfactory intraobserver reproducibility, did not meet the criteria of a reliable prognostic factor. Still, the ISUP/WHO scale has only been validated for clear cell RCC and papillary RCC because there have not been enough cases of other subtypes. It is important to mention that there are subtypes of RCC in the appearance of nucleoli does not correlate with the overall grade and prognosis and should be used for descriptive purposes only [4, 8].
Other changes in current classification include an increase of the maximal diameter of papillary adenoma up to 1,5 cm (and increasing the pool of possible kidney donors). Adult cystic nephroma was shifted to mixed epithelial-stromal tumours as a part of their spectrum. Moreover, the term “renal carcinoid” was replaced with “well-differentiated neuroendocrine tumour of the kidney” in order to emphasize the metastatic changes after nephrectomy and poor prognosis. Similar principles led to changing the name of “multilocular cystic renal cell carcinoma” to “multilocular cystic renal neoplasm of low malignant potential” to highlight the lack of malignant potential. Papillary RCC type 2 is now believed to be more than one type of tumour as its subtypes contain different molecular changes [7].
Presented cases
Acquired cystic disease (ACD)-associated RCC is a subtype newly included in the 2016 WHO classification. It was first described in 2005 and it was recognized in the 2013 ISUP Vancouver classification of renal neoplasia [9]. This tumour arises in patients with acquired cystic kidney disease due to end-stage renal disease, often in association with long-term haemodialysis or peritoneal dialysis [4, 9]. It is now the most common subtype of RCC in patients with end-stage renal disease and ACD and accounts for over 1/3 of renal tumours in these patients [4].
Patients are usually asymptomatic and the tumour is found accidentally during routine check-up and imaging [9, 10]. This tumour occurs predominantly in male patients with ACD of younger age but the incidence of carcinoma increases with duration of dialysis [9-11], recent studies report that it is most likely to be diagnosed between 10 to 20 years after the first dialysis [9, 12]. The risk is also increased for individuals with acquired cystic kidney disease who underwent renal transplantation [5].
The ACD-associated RCC tumour is usually well-circumscribed, cystic and/or solid. The cut surface varies from yellow-tan to brown, focal necrosis and haemorrhage may be present [9, 13]. Histologically, these tumours are composed of cells with abundant eosinophilic cytoplasm and large irregular vesicular nuclei with prominent nucleoli. They show a broad spectrum of architectural intermixed patterns including papillary, tubular, acinar and solid, however sarcomatoid and/ or rhabdoid features have also been described [13-15]. Another common feature characteristic of this RCC is the presence of intracytoplasmatic and/or intercytopasmatic lumina and intratumoral calcium oxalate crystal deposition [1, 4, 9-10]. Due to its unique morphological features, immunohistochemistry is not required to diagnose ACD-RCC. The most common pattern is positivity for CD10, RCC marker, AMACR. CK 7 is typically not expressed or expressed focally and CAIX is not detected [1, 8-9]. The genetic alterations in ACD-associated RCC are quite complex and even samples taken from the same tumour may not be genetically identical. The most common abnormalities are gains in chromosomes 3, 7, 16 [10]. Mutations in von Hippel-Lindau (VHL) gene have not been reported [5]. In general, ACD-associated RCC seems to be less aggressive than sporadically occurring RCC and has a better prognosis [9]. Most tumours have indolent behaviour, although some patients may develop metastatic disease [4, 8, 10]. However, it is worth remembering that almost any histological variant of RCC can occur in patients with an acquired cystic disease, but the most common types are ACD-associated RCC and papillary RCC [9]. ACD-RCC might be misdiagnosed as type 2 papillary RCC because its papillary architectural pattern and the presence of clear cell areas may lead to confusion with classical clear cell RCC [3]. The morphology of the presented case, especially combination of multiple architectural patterns, characteristic lumina, as well pathological changes in surrounding kidney together with patient’s clinical history formed our diagnosis.
Tubulocystic renal cell carcinoma is an uncommon (<1% of all RCC) renal epithelial malignancy [16]. It has male predominance (7:1), patients’ age ranges from 30 to 94 years [17-18]. Majority of tubulocystic RCCs are discovered incidentally, but patients may present symptoms such as abdominal pain and hematuria [19]. In rare instances, it may occur in patients with existing end-stage renal disease [1, 17-18]. On macroscopic examination, this tumours tends to be a solitary, well-circumscribed multicystic renal mass, with a mean diameter of about 4 cm [20]. Cysts are numerous, small to intermediate in size with spongy cut surface [17-18]. Cysts are lined by single layer of flattened, cuboidal or columnar as well as hobnail epithelium. The neoplastic cells nuclei are enlarged and irregular with intermediate to large nucleoli [19, 21]. Enlarged nucleoli are one of the diagnostic features of tubulocystic RCC, but nuclear grading should not be applied as it does not correlate with the outcome [18, 20]. The cytoplasm is abundant and sometimes eosinophilic. Cyst and tubules are separated by thin fibrovascular septa. There may be also components similar to papillary RCC.
Immunohistochemically, tubulocystic carcinoma has similar features to papillary RCC and shows positive staining for racemase, CK7, CD10, and RCC antigen [1, 16-17]. The genetic studies of tubulocystic RCC ale limited and they show some overlapping features with papillary RCC [15]. The most commonly reported were gains of chromosomes 7 and 17 and loss of the Y chromosome [1, 5]. Majority of tubulocystic RCC have indolent behaviour with very few recurrences and unusual metastases [5, 11, 20]. The differential diagnosis includes other tumours with a multiloculated gross appearance: multilocular cystic RCC (multilocular cystic renal neoplasm of low malignant potential), cystic nephroma, mixed epithelial and stromal tumours, cystic oncocytoma [16, 18]. The diagnosis in our case was quite problematic as it demanded a careful examination of tumour architectural patterns and cytology. Its cells exhibited ISUP grade 2 nucleoli, whereas according to WHO recommendations they should be ISUP grade 3. Therefore, immunohistochemistry and especially the tumor’s overall benign appearance were helpful in making the final diagnosis.
The last of our cases presented the biggest diagnostic challenges. There are only several published cases of the unique renal neoplasm characterized by eosinophilic cytoplasm and solid and cystic growth reported in patients with tuberous sclerosis complex (TSC) [7, 22]. TSC is an autosomal dominant disorder with characteristic tumours and tumor-like conditions involving multiple organs, while in the kidney the most common tumour is angiomyolipoma [23]. RCC is less frequently reported in this syndrome, but may have very distinct morphology. Some of the RCCs coexisting with TSC show features similar to chromophobe RCC or are described as RCC with smooth muscle stroma, but there are several reports of RCC with a granular eosinophilic-macrocystic morphology [23]. These tumours showed female predominance and occurred at a younger age. Recently, eosinophilic solid and cystic RCC has been documented also in the series of female patients without clinical features of TSC [7]. These tumours, usually asymptomatic, well-defined, were located in the medulla. Cut surface was tan with typically large macrocytic spaces, variable in size, interspersed with solid nodules [7].
Microscopically they contain solid areas admixed with variably sized macrocysts and microcysts lined by cells with abundant eosinophilic cytoplasm and hobnail arrangement. Nuclei are round to oval with prominent nucleoli. There is often chronic inflammatory infiltrate with multinucleated cells within the neoplastic stroma. Some cases have a predominantly microcystic arrangement or septa compressed between solid nodules and therefore difficult to spot [7, 22]. Immunoprofile shows nuclear PAX8 expression, predominant CK20-positive/ CK7-negative phenotype (but CK20-positive/CK7-positive and CK20-negative/CK7-negative phenotypes exist), patchy AMACR staining, vimentin usually positive, CD10 focally positive and CAIX negative in most cases [7]. This tumour is believed to have indolent behaviour, but data is still lacking. There is a discussion whether to even label it as “of uncertain malignant potential” or “renal cell carcinoma.” So far these types of tumours were described mainly as „unclassified," however it is important to recognize them in order to determine their true biology [7]. It was those peculiar cytological features, especially eosinophilia and multinuclearity, that caught our attention and pushed toward correct diagnosis after the literature review.
Renal cell carcinoma, subtype eosinophilic solid and cystic are among the emerging entities, not included in the current classification, but candidates for the future because of their distinctive morphology and immunohistochemical features [7].
Conclusion
This report shows that new entities recognised by 2016 WHO Classification of Tumors of the Urinary System can be found in everyday practice based on careful morphological assessment [5]. Therefore it is important to be able to diagnose such tumours and then classify them according to the latest guidelines and in correlation to patient's history [24]. It is also important to remember that classification of renal cell carcinoma is still evolving and new tumours with unique morphological, immunohistochemical and molecular patterns are constantly reported and may emerge as a distinctive subtype in future.
References
| 1. |
Delahunt B, Srigley JR. The evolving classification of renal cell neoplasia. Semin Diagn Pathol. 2015;32(2):90-102.
|
| 2. |
Nouh MA, Kuroda N, Yamashita M, et al. Renal cell carcinoma in patients with end-stage renal disease: relationship between histological type and duration of dialysis. BJU Int. 2010;105(5):620-7.
|
| 3. |
Nguyen DP, Vertosick EA, Corradi RB, et al. Histological subtype of renal cell carcinoma significantly impacts survival in the era of partial nephrectomy. Urol Oncol. 2016;34(6):259.e1-259.e8.
|
| 4. |
Udager AM, Mehra R. Morphologic, Molecular, and Taxonomic Evolution of Renal Cell Carcinoma: A Conceptual Perspective With Emphasis on Updates to the 2016 World Health Organization Classification. Arch Pathol Lab Med. 2016;140(10):1026-37.
|
| 5. |
Moch H, Humphrey PA, Ulbright TM, Reuter VE, eds. WHO Classification of Tumors of the Urinary System and Male Genital Organs. 4th ed. Lyon, France: IARC; 2016. World Health Organization Classification of Tumors; vol 8.
|
| 6. |
Brierley J, Gospodarowicz M, Wittekind C., editors. UICC TNM classification of malignant tumours. Eighth ed, Wiley; 2017. p 199-201.
|
| 7. |
Trpkov K, Hes O, Bonert M, et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am J Surg Pathol. 2016;40(1):60-71.
|
| 8. |
Moch H, Cubilla AL, Humphrey PA, et al. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur Urol. 2016;70(1):93-105. doi: 10.1016/j.eururo.2016.02.029.
|
| 9. |
Foshat M, Eyzaguirre E. Acquired Cystic Disease-Associated Renal Cell Carcinoma: Review of Pathogenesis, Morphology, Ancillary Tests, and Clinical Features. Arch Pathol Lab Med. 2017;141(4):600-606.
|
| 10. |
Bhatnagar R, Alexiev BA. Renal-cell carcinomas in end-stage kidneys: a clinicopathological study with emphasis on clear-cell papillary renal-cell carcinoma and acquired cystic kidney disease-associated carcinoma. Int J Surg Pathol. 2012;20(1):19-28.
|
| 11. |
Crumley SM, Divatia M, Truong L, et al. Renal cell carcinoma: Evolving and emerging subtypes. World J Clin Cases. 2013; 1(9): 262-275.
|
| 12. |
Bonsib SM. Renal cystic diseases and renal neoplasms: a mini-review. Clin J Am Soc Nephrol. 2009;4(12):1998-2007. doi: 10.2215/CJN.02020309.
|
| 13. |
Kuroda N, Naroda T, Tamura M, et al. Acquired cystic disease-associated renal cell carcinoma: a clinicopathological study of seven cases. Pol J Pathol. 2017;68(4):306-311.
|
| 14. |
Tickoo SK, dePeralta-Venturina MN, Harik LR, et al. Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol. 2006;30(2):141-53.
|
| 15. |
Rao Q, Xia Q-Y, Cheng L, et al. Molecular genetics and immunohistochemistry characterization of uncommon and recently described renal cell carcinomas. Chin J Cancer Res. 2016;28(1):29-49.
|
| 16. |
Sarungbam J, Mehra R, Tomlins SA, et al. Tubulocystic renal cell carcinoma: a distinct clinicopathologic entity with a characteristic genomic profile. Mod Pathol. 2019 Jan 8.
|
| 17. |
Kryvenko ON, Jorda M, Argani P, et al. Diagnostic approach to eosinophilic renal neoplasms. Arch Pathol Lab Med. 2014;138(11):1531-41.
|
| 18. |
Kuroda N, Matsumoto H, Ohe C, et al. Review of tubulocystic carcinoma of the kidney with focus on clinical and pathobiological aspects. Pol J Pathol. 2013;64(4):233-7.
|
| 19. |
Banerjee I, Yadav SS, Tomar V, Yadav S, Talreja S. Tubulocystic Renal Cell Carcinoma: A Great Imitator. Rev Urol. 2016;18(2):118-121.
|
| 20. |
Mehra R, Smith SC, Divatia M, et al. Emerging Entities in Renal Neoplasia. Surg Pathol Clin. 2015;8(4):623-56.
|
| 21. |
Hora M, Urge T, Eret V, et al. Tubulocystic renal carcinoma: a clinical perspective. World J Urol. 2011;29(3):349-54.
|
| 22. |
Moch H, Ohashi R, Gandhi JS. Et al. Morphological clues to the appropriate recognition of hereditary renal neoplasms. Semin Dign Pathol. 2018;35(3):184-192.
|
| 23. |
Guo J, Tretiakova MS, Troxell ML, et al. Tuberous sclerosis-associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014;38(11):1457-67.
|
| 24. |
Delahunt B, Eble JN, Egevad L, et al. Grading of renal cell carcinoma. Histopathology. 2019;74(1):4-17.
|
